Antes de mostrar o desenvolvimento de um programa para aquisição e armazenamento das leituras de um medidor multiparâmetro (OD, pH e Condutividade) resolvi organizar algumas informações sobre os fundamentos teóricos envolvidos em duas técnicas instrumentais usadas para o monitoramento de OD, sensor eletroquímico e sensor óptico.
Para quem já conhece (ou não tem interesse, ou tempo, para conhecer) os fundamentos dos sensores eletroquímicos e ópticos de OD, pode ir direto para a próxima seção.
O monitoramento de OD é importante em todos os processos onde a concentração de oxigênio influencia a velocidade das reações, a eficiência do processo ou as condições ambientais. Algumas aplicações importantes incluem o tratamento de efluentes, processos fermentativos, bioprocessos em geral e o monitoramento hídrico ambiental.
O monitoramento da concentração de Oxigênio Dissolvido é uma ferramenta muito importante em qualquer laboratório de águas. Para saber mais sobre a importância do Oxigênio Dissolvido leia o ítem “Reações Redox em Água” disponível na artigo Análise da Água.
Sugiro também como fonte de consulta para este assunto a Tese de Doutoramento: “Desenvolvimento de sensores de oxigênio dissolvido utilizando métodos eletroquímicos e ópticos para monitoramento em tempo real da qualidade da água”(Ferreira M.A.C., 2007).
Existem dois tipos de técnicas eletroquímicas para medir O2 em solução: eletrodo galvânico e eletrodo polarográfico. [6] Em ambos os casos utiliza-se um eletrodo que reage com o O2 dissolvido gerando uma corrente que é proporcional à quantidade de O2 consumido.
Em um sistema galvânico os materiais do eletrodo são selecionados de modo a gerar uma diferença de potencial entre o cátodo e o ânodo maior ou igual a -0,5 volts dispensando a aplicação de um potencial externo. Enquanto que no sistema polarográfico é necessário a aplicação de uma voltagem externa. (Fonte: www.sensorex.com)
O eletrodo polarográfico comumente usado para medir OD é também chamado de eletrodo de Clark em homenagem ao seu idealizador, Leland C. Clark Jr. (1918-2005), e usado inicialmente para medir a concentração de O2 no sangue durante cirurgias cardíacas.
O eletrodo de Clark é uma célula polarográfica formada por um cátodo e um ânodo, com a aplicação de um potencial de ~800mV, em uma solução de eletrólito e separados do meio externo por uma membrana permeável a gás, geralmente Teflon™.
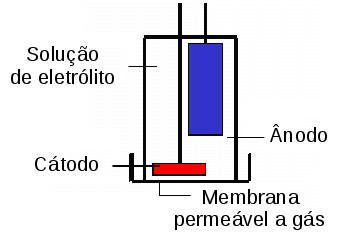
Para o cátodo é utilizado um metal nobre (ouro, prata ou platina) e como ânodo utiliza-se um eletrodo de prata/cloreto de prata (Ag/AgCl), ambos mergulhados em uma solução de eletrólito de cloreto de potássio (KCl - 0,1M) ou brometo de potássio (KBr).
O oxigênio é reduzido no cátodo formando íons hidróxido:
4e- + O2 + 2H2O -> 4OH-
E os elétrons para essa reação são fornecidos pela oxidação do eletrodo de Ag:
4Ag + 4Cl- -> 4AgCl + 4e-
O consumo de O2 gera um diferencial de pressão parcial de O2 entre o interior do eletrodo e o meio externo, favorecendo a difusão de O2 através da membrana do meio externo para a solução de eletrólito.
Portanto, para cada de molécula de O2 reduzida no cátodo são liberados 4 elétrons pelo ânodo gerando uma corrente proporcional ao O2 consumido.
Com a oxidação do eletrodo de prata forma-se, ao longo do tempo, um precipitado de AgCl na superfície do ânodo reduzindo a área de contato do ânodo com a solução.
Quando o ânodo está completamente recoberto a reação é interrompida e o eletrodo para de funcionar. Para ser reativado o eletrodo deve ser desmontado para remoção da camada de AgCl da superfície do ânodo.
Esta operação demanda tempo e implica em interrupção no monitoramento.
A formação de íons hidróxido no cátodo aumenta o pH do meio e desloca as leituras do zero, além disso o consumo de íons cloreto no ânodo também contribui para alterar a composição da solução por isso o eletrólito deve ser trocada conforme o uso do eletrodo (de duas semanas a 6 meses).
A quantificação de OD depende da difusão das moléculas de O2 através da membrana e portanto qualquer modificação da permeabilidade da membrana interfere nas medidas.
Um dos possíveis problemas é a mudança da permeabilidade pela formação de biofilmes bacterianos na superfície da membrana, e o consumo de O2 por organismos presentes no biofilme.
O aumento de temperatura da membrana também aumenta a sua permeabilidade e por isso os equipamentos costumam incluir um sensor de temperatura (termistor) da membrana para fazer a devida compensação das leituras.
Com a difusão de O2 através da membrana a concentração de O2 no região próxima da membrana fica menor do que no restante da solução e a leitura fica dependente da difusão das moléculas de O2 na água. Para compensar este efeito a amostra é mantida sob agitação durante as medidas para garantir a homogeneidade e evitar a formação de gradientes de concentração entre a superfície da membrana e o restante da solução.
Quando um metal entra em contato com uma solução, forma-se uma dupla camada elétrica devido à separação de cargas na interface metal-solução, criando uma diferença de potencial através da interface. Em condicões de equilíbrio, não há fluxo de cargas e a diferença de potencial da interface é Eeq. Experimentalmente este valor (Eeq) não é mensurável. Quando este equilíbrio é perturbado, de forma espontânea ou induzida, ocorrem transferências de elétrons através da interface. E esse deslocamento do equilíbrio é chamado de Polarização. Quando um eletrodo está nestas condições diz-se que está Polarizado. (Fonte: http://electrochemistry.wordpress.com)
A Polarização pode ser espontânea, como nos processos corrosivos, ou induzida como em um sensor polarográfico para OD.
Quando o medidor é ligado é aplicado um potencial ao eletrodo de ~800mV e deve-se aguardar um intervalo de 5 a 25 min de Polarização, para estabilizar a corrente, antes de fazer uma leitura.
O manual dos sensores de OD da Thermo recomenda que uma sonda nova deve ser polarizada por um período de 30 a 60 minutos. De acordo com o manual, o sensor de OD está continuamente polarizado enquanto o medidor estiver ligado mas se o medidor for desligado por um período menor que 1 hora o sensor deve ser novamente polarizado por um período de 5 a 25 minutos.
Existem três alternativas para a calibração de sensores de OD. (Fonte: www.coastalwiki.org/coastalwiki/Oxygen_sensors
Comparação com amostras de água fatoradas pelo método Winkler.
Este é o método que oferece maior exatidão no entanto é um procedimento trabalhoso e exige experiência.
Calibração em “água” saturada de “ar”.
Este método está sujeito a erros devido à possibilidade de supersaturação da água, além de exigir agitação e aeração constante durante a calibração.
Calibração em “ar” saturado de “água”.
Esta calibração é feita com o sensor em um recipiente pequeno semi-fechado (pressão interna deve ser igual à pressão externa), com uma esponja molhada na parte inferior.
Deve-se evitar variações de temperatura durante a calibração e evitar a presença de gotas de água na superfície da membrana
Segundo o guia do usuário dos sensores de OD da Thermo a calibração pode ser feita rapidamente pelo método do “ar” saturado de “água”.
Este método é possível pois a pressão parcial de O2 em “água” saturada de “ar” é igual à pressão parcial de O2 no “ar” saturado de “água” (ar com 100% de umidade relativa).
Para entender melhor esse conceito consulte o apêndice: Solução de Gás em Líquido.
Isso significa que uma sonda calibrada em “ar” saturado de “água” será capaz de medir a pressão parcial de oxigênio em uma amostra de água.
Como a taxa de difusão do O2 na água é ligeiramente diferente da taxa de difusão no ar o medidor aplica um fator de correção de 102,3% para a calibração feita no “ar” saturado de “água” para obter o valor correspondente à “água” saturada de “ar”.
O manual também recomenda que para medidas de OD menores que 2 ppm é recomendável um segundo ponto de calibração com um padrão isento de OD.
Os sensores ópticos para OD, também chamados de “optodos” ou “optrodos”, se baseiam na capacidade do Oxigênio de suprimir a Fluorescência emitida por um Fluoróforo. E portanto a intensidade do efeito de supressão da fluorescência pode ser correlacionada com a concentração do Oxigênio.
Mas antes vamos ver um pouco de teoria para melhor entendermos os fenômenos de Fluorescência e Supressão da Fluorescência.
Utilizamos com principais fontes de consulta para esta seção os livros Skoog, 2002 e Lakowicz, 2006, os sites www.fisicaequimica.net e http://micro.magnet.fsu.edu, e as teses “Optodos para a Determinação de SO2 e O2” (Silva K.R.B., 2007), “Desenvolvimento de sensores de Oxigênio Dissolvido utilizando métodos eletroquímicos e ópticos para monitoramento em tempo real da qualidade da água” (Ferreira M.A.C., 2007) e “Investigação de Carboidratos Anfifílicos com Micelas de SDS e Lipossomos de DPPC” (Nei M.C.G., 1997)
A associação de átomos para a formação de moléculas é um processo energeticamente favorável pois permite equilibrar as forças de repulsão (elétron <> elétron e núcleo <> núcleo ) com as forças de atração (elétron >< núcleo).
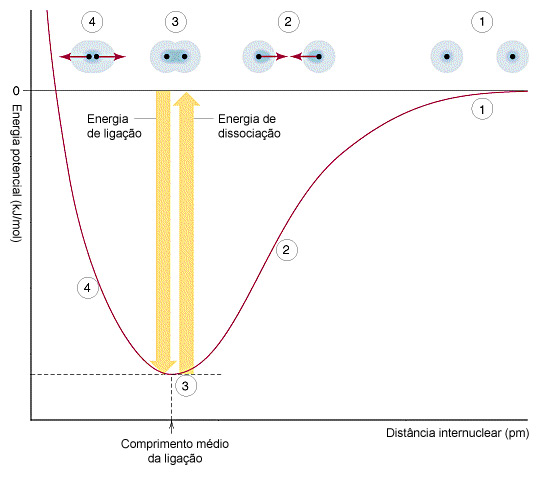
Um gráfico da energia de ligação de uma molécula diatômica (Ex: O2, Cl2, HCl) em função da distância internuclear [7] mostra uma região de energia mínima onde as forças de atração e repulsão estão em equilíbrio.
Figura 136. Curva de energia potencial da ligação de uma molécula diatômica em função da distância internuclear. (Fonte: www.fisicaequimica.net)
Uma molécula, após a sua formação, não permanece indefinidamente neste “poço” de energia potencial, mas pode absorver ou liberar energia passando para diferentes estados energéticos devido à absorção ou emissão de radiação pelos elétrons.
A Teoria Quântica é a ferramenta teórica usada para explicar os processos de absorção e liberação de energia.
A Teoria Quântica foi primeiramente proposta em 1900 por Max Planck, um físico alemão, para explicar as propriedades de radiação emitida por corpos aquecidos e foi posteriormente estendida para racionalizar outros tipos de processos de emissão e absorção.
A teoria quântica inclui dois postulados importantes:
Átomos, íons ou moléculas podem existir somente em certos estados discretos, caracterizados por quantidades definidas de energia. Quando uma espécie altera seu estado, absorve ou emite uma quantidade de energia “exatamente” igual à diferença de energia entre os estados.
Quando átomos, íons ou moléculas absorvem ou emitem radiação ao efetuar uma transição de um estado de energia para outro, a radiação de frequência ν ou de comprimento de onda λ está relacionada com a diferença de energia entre os dois estados pela equação:
E1 - E0 = hν = hc/λ
Onde E1 é a energia do estado mais alto, E0 é a energia do estado mais baixo, c é a velocidade da luz e h é a constante de Planck.
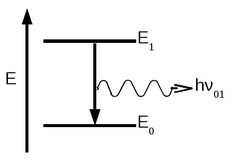
O estado de menor energia de um átomo ou molécula é chamado de estado fundamental e os estados de mais alta energia são chamados de estados excitados.
Uma importante contribuição para o conteúdo energético de átomos ou íons é a energia associada ao movimento dos elétrons em torno do núcleo positivamente carregado. Por isso os diferentes estados energéticos de átomos ou íons são também chamados de estados eletrônicos.
As moléculas, além de apresentarem diferentes estados eletrônicos, também apresentam diferentes estados vibracionais quantizados associados à energia das vibrações interatômicas e diversos estados rotacionais quantizados associados à energia das rotações das moléculas em torno de seus eixos de rotação.
Nota
À temperatura ambiente, praticamente todas as moléculas em uma solução estão no mais baixo nível vibracional do estado fundamental e portanto usualmente a excitação ocorre a partir deste nível de energia.
Portanto a energia “E” associada às bandas do espectro de uma molécula é constituída por três componentes:
E = Eeletrônica + Evibracional + Erotacional
Eeletrônica descreve a contribuição da energia resultante dos estados de energia de todos os elétrons das ligações.
Evibracional se refere à contribuição de todos os tipos de possíveis vibrações interatômicas presentes na molécula
Erotacional é a energia resultante dos vários movimentos rotacionais presentes na molécula.
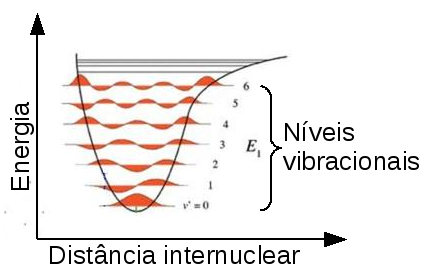
Geralmente existem muitos níveis de energia vibracional quantizada em cada nível de energia eletrônico quantizado, conforme ilustra a figura seguinte.
Figura 140. Diferentes níveis de energia vibracional que uma molécula pode assumir em um mesmo estado eletrônico. Estão omitidos, por simplicidade, os níveis rotacionais associados a cada nível vibracional. (Fonte: http://en.wikipedia.org/wiki/Franck-Condon_principle)
Existem muitos níveis de energia rotacionais associados a cada nível vibracional, pois a diferença de energia entre os níveis de energia rotacionais é pequena relativamente às diferenças de energia entre os níveis vibracionais. Transições rotacionais ocorrem na região de microondas e infravermelho distante (comprimentos de onda de 0,01 a 1 cm).
As moléculas podem sofrer transições vibracionais “puras” pela absorção ou emissão de um fóton com energia na região do Infravermelho, ou seja de 0,78 μm a 1000 μm, faixa de energia insuficiente para provocar transições eletrônicas.
A absorção, ou emissão, de fótons na região do UV/Vis [8] provoca transições nos níveis de energia eletrônicos e vibracionais de uma molécula, e por isso são chamadas transições vibrônicas.
Nota
Radiação ultravioleta de comprimentos de onda menores que 250 nm é suficiente energética para causar ruptura de ligações e provocar reações fotoquímicas.
A figura seguinte ilustra uma transição vibrônica hipotética entre dois níveis eletrônicos, E0 e E1.
Figura 141. Transição vibrônica hipotética do subnível vibracional ν = 0 do nível eletrônico E0 para o subnível vibracional ν = 2 do nível eletrônico E1 com absorção de energia, indicada pela seta de cor azul, e decaimento do subnível vibracional ν = 0 do nível eletrônico E1 para o subnível vibracional ν = 2 do nível eletrônico E0 com emissão de energia, indicada pela seta de cor verde. (Fonte: http://en.wikipedia.org/wiki/Franck-Condon_principle)
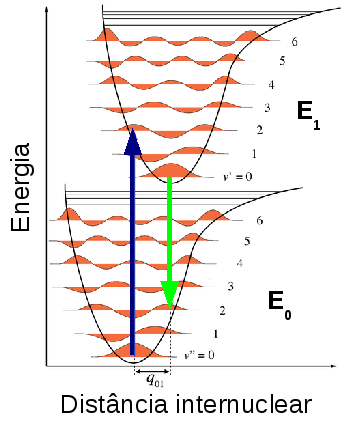
As transições entre os estados são representados como linhas verticais para ilustrar a natureza instantânea de absorção da luz, na ordem de 10-15 s, durante a qual não ocorre deslocamento significativo da distância entre os núcleos, o que é conhecido por Princípio de Franck-Condon.
Após essas informações básicas sobre os níveis energéticos das moléculas vamos recapitular também os conceitos de Spin Eletrônico e os estados Singlete, Dublete e Triplete.
O princípio de exclusão de Pauli diz que dois elétrons em um átomo não podem ter os mesmos valores dos quatro números quânticos (n, l, ml e ms). Essa restrição exige que apenas dois elétrons podem ocupar um mesmo orbital e além disso os dois devem ter spin [9] opostos. Nessa circunstância, diz-se que os spins estão emparelhados.
Por isso a maioria das moléculas não apresenta campo magnético intrínseco e são chamadas de diamagnéticas, ou seja, não são atraídas nem repelidas por campos magnéticos estáticos.
Por isso os radicais livres são paramagnéticos, ou seja, são atraídos por um campo magnético pois possuem elétrons desemparelhados e por isso apresentam um momento magnético.
Uma molécula na qual todos os spins eletrônicos estão emparelhados é classificada como uma molécula no estado singlete porque nesse estado não ocorre separação dos níveis de energia quando a molécula é exposta a um campo magnético.
Mas o estado fundamental de um radical livre é chamado de estado dublete, porque o elétron desemparelhado pode assumir duas orientações diferentes em um campo magnético correspondendo a diferentes níveis de energia.
Quando um elétron de uma molécula no estado fundamental singlete absorve energia e é excitado para um orbital em um nível mais alto de energia pode-se formar um estado singlete ou triplete, conforme a figura seguinte.
Figura 142. Representação esquemática dos estados fundamental e excitado, singlete e triplete (multiplicidade do spin) com as setas indicando as orientações de spin.
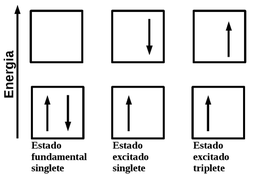
No estado excitado singlete, o spin do elétron promovido, a um nível de maior energia, ainda está emparelhado com o elétron no estado fundamental. Enquanto que no estado triplete os spins dos dois elétrons ficaram desemparelhados ou paralelos.
O estado excitado triplete tem energia menor que o estado singlete correspondente.
A absorção de um fóton de energia ocorre devido a uma interação do vetor campo elétrico oscilante da onda de luz com as cargas (elétrons) da molécula.
Se o fóton absorvido contém mais energia do que é necessário para uma transição eletrônica simples, o excesso de energia geralmente é convertida em energia vibracional e rotacional, pois existem numerosos níveis vibracionais para cada nível eletrônico assim como muitos níveis rotacionais para cada nível vibracional.
Nota
Mas lembrar que para que a absorção da radiação ocorra, a energia do fóton de excitação deve ser exatamente igual à diferença de energia entre o estado fundamental e um estado excitado na amostra absorvedora.
No entanto, caso ocorra uma colisão entre uma molécula e um fóton com energia insuficiente para promover uma transição, não ocorre absorção.
Com as informações básicas sobre níveis energéticos de moléculas e os conceitos de spin, singlete, dublete e triplete vamos passar para o fenômeno da Fluorescência.
A "fluorescência", juntamente com a “fosforescência” e a “quimiluminescência”, é um dos três tipos de métodos ópticos de “Luminescência” onde moléculas (ou átomos) são excitados para níveis mais altos de energia e decaem posteriormente para níveis de menor energia podendo emitir fótons cujo espectro de emissão pode fornecer informações qualitativas ou quantitativas sobre as moléculas (ou átomos) e a sua vizinhança.
A fluorescência e fosforescência são semelhantes pois em ambas a excitação ocorre pela absorção de fótons, enquanto que na quimiluminescência a espécie excitada é formado por uma reação química.
A fluorescência ocorre em sistemas químicos gasosos, líquidos e sólidos. O exemplo mais simples de fluorescência é o que ocorre com vapores atômicos diluídos. [10] Por exemplo os elétrons do orbital 3s de átomos de vapor de sódio podem ser excitados ao orbital 3p por absorção de radiação de comprimento de onda de 5.89 e 5.896 Å (ou 589,0 e 589,6 nm)(Ver: Absorção da Radiação).
Após 10-5 a 10-8 s, os elétrons voltam ao estado fundamental e, ao fazer isso, emitem radiação dos mesmos dois comprimentos de onda (5.896 e 5.890 Å) em todas as direções. Esse tipo de fluorescência, em que a radiação absorvida é reemitida sem mudança de frequência , é conhecida como radiação de ressonância ou fluorescência de ressonância.
Além dos átomos muitas moléculas também exibem fluorescência de ressonância, no entanto é mais frequente a emissão da radiação de fluorescência em comprimentos de onda maiores (menor energia) do que a radiação absorvida, o que é conhecido por deslocamento Stokes.
A figura seguinte, chamada de Diagrama de Jablonski [11] , mostra um diagrama parcial de níveis de energia de uma molécula fotoluminescente típica. A linha horizontal grossa mais inferior representa a energia do estado fundamental da molécula, que é normalmente um estado singlete (S0).
Nota
À “temperatura ambiente”, praticamente todas as moléculas em uma solução estão no mais baixo nível vibracional do estado fundamental.
As linhas grossas superiores são os níveis de energia para os estados fundamentais vibracionais de três estados eletrônicos excitados (S1, S2 e T1).
Figura 143. Diagrama parcial de níveis de energia de uma molécula fotoluminescente. (Fonte: www.monografias.com
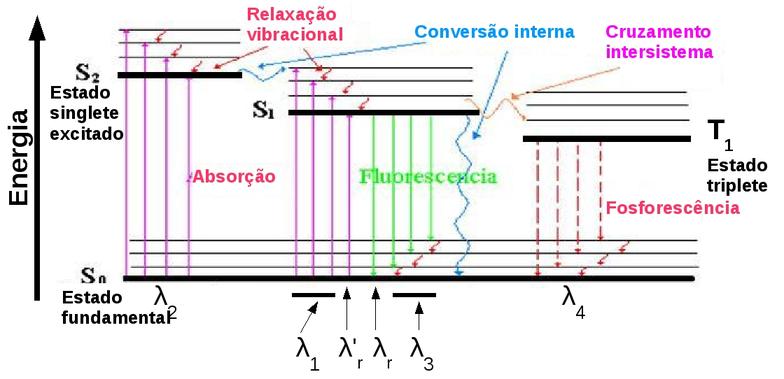
As duas linhas à esquerda representam o primeiro (S1) e segundo (S2) estados eletrônicos excitados singletes.
A da direita (T1) representa a energia do primeiro estado eletrônico triplete. Como é normalmente o caso, a energia do primeiro estado excitado triplete é menor que a energia do estado singlete correspondente.
As linhas horizontais mais finas indicam os níveis vibracionais associados a cada um dos quatro estados eletrônicos (S0, S1, S2 e T1)
A excitação pode ocorrer pela absorção de radiação em duas regiões distintas centradas em λ1 (S0 -> S1) e λ2 (S0 -> S2).
A absorção em λ1 (S0 -> S1) é de menor energia do que a transição em λ2 (S0 -> S2) e portanto λ1 tem maior comprimento de onda do que λ2.
Observe que nos processos de excitação (S0 -> S1) e (S0 -> S2) a molécula pode ocupar diversos estados excitados vibracionais.
Observe também que a excitação direta (S0 -> T1) não está mostrada pois é um evento com baixa probabilidade de ocorrência e por isso é chamada de transição proibida.
A irradiação de uma substância com um amplo espectro de comprimentos de onda irá ocasionar uma ampla gama de transições permitidas levando as moléculas para diferentes níveis de energia vibracional do estado excitado. Mas algumas transições têm maior probabilidade de ocorrência do que outras, que quando combinadas, formam o perfil do espectro de absorção.
Na maioria dos casos os espectros de absorção e emissão são distintos, mas frequentemente ocorre a sobreposição parcial e algumas vezes a sobreposição pode ser completa.
Figura 144. Espectro hipotético de absorção, emissão e a região de sobreposição parcial. (Fonte: http://micro.magnet.fsu.edu
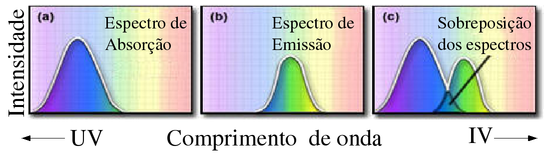
A fluorescência e a fosforescência são mais facilmente observáveis a um ângulo de 90 graus em relação ao feixe incidente.
Uma molécula excitada pode voltar ao estado fundamental através de vários processos com diferentes probabilidades de ocorrência.
Conforme mostrado no diagrama de Jablonski, os processos de fluorescência e fosforescência envolvem a emissão de um fóton de radiação nas bandas λ3 e λ4 respectivamente.
Os outros processos de desativação, indicados por setas sinuosas (relaxação vibracional, conversão interna e cruzamento intersistema), são processos não-radiativos.
Conforme o diagrama de Jablonski uma molécula pode ser levada a qualquer um dos vários níveis vibracionais durante o processo de excitação eletrônica. Mas, em solução, a energia vibracional em excesso é perdida imediatamente pelas colisões entre a molécula excitada e o solvente, num processo chamado relaxação vibracional.
Por isso, a fluorescência de uma solução envolve a transição a partir do nível vibracional mais baixo de um estado eletrônico excitado para qualquer um dos níveis vibracionais do estado fundamental, a partir dos quais ela cairá rapidamente ao nível vibracional mais baixo do estado fundamental por relaxação vibracional subsequente.
A conversão interna é outro processo de desativação pelo qual a moléculas excitada passa para “outro” estado eletrônico de menor energia sem emissão de radiação.
A conversão interna parece ser mais eficiente quando os dois níveis eletrônicos envolvidos estão próximos o suficiente para que haja uma superposição de níveis de energia vibracionais, conforme está representado na conversão interna S2 -> S1 do diagrama de Jablonski.
A conversão interna S2 -> S1 ilustrada no diagrama de Jablonski acima explica porque se obtém uma banda de fluorescência somente em λ3, não importando se a excitação foi feita no comprimento de onda λ2 ou λ1.
O Quinino é um exemplo clássico desse tipo de comportamento, conforme ilustrado na figura seguinte.
Figura 145. Espectros de excitacão e emissão de fluorescência de uma solução de quinino.(Fonte: www.rose-hulman.edu
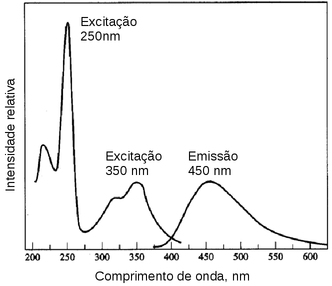
O quinino tem duas bandas de excitação em 250 nm e 350 nm, mas independente do comprimento usado para excitar a molécula o espectro de emissão de fluorescência tem um máximo em 450 nm.
No entanto os mecanismos envolvidos na conversão interna S1 -> S0 (decaimento não radiativo), indicada no diagrama de Jablonski acima, ainda não são bem compreendidos.
O cruzamento intersistema é um processo no qual o spin de um elétron excitado é invertido resultando uma mudança na multiplicidade da molécula.
Da mesma forma que na conversão interna, a probabilidade desse tipo de transição é aumentada se os níveis vibracionais dos dois estados se interpenetram.
Na transição singlete/triplete (S1 -> T1) indicada no diagrama de Jablonski o estado vibracional inferior do singlete se superpõe a um dos níveis vibracionais superiores do triplete, aumentando a probabilidade de uma mudança no estado do spin.
O cruzamento intersistema é mais comum em moléculas que contêm átomos pesados, como iodo ou bromo (efeito do átomo pesado).
A presença de espécies paramagnéticas, tal como o oxigênio molecular, na solução também facilita o cruzamento intersistema e um consequente decréscimo da fluorescência.
A desativação de um estado eletrônico excitado pode envolver a interação e transferência de energia entre a molécula excitada e o solvente ou outros solutos. Esses processos são chamados coletivamente conversão externa, ou supressão (ou extinção por colisão).
A ocorrência de conversão externa pode ser indicada quando há marcante efeito de solvente na intensidade de fluorescência.
As condições que tendem a reduzir o número de colisões entre partículas (baixa temperatura e alta viscosidade) geralmente aumentam a fluorescência.
Mas os detalhes dos processos de conversão externa também não são bem-compreendidos!
A fosforescência é um mecanismo alternativo, luminescente, para a desativação de estados eletrônicos excitados.
Depois do cruzamento intersistema para um estado triplete, uma desativação subsequente pode ocorrer, seja por conversão interna, externa ou por fosforescência.
Uma transição triplete -> singlete (Ex: T1 -> S0) é muito menos provável que uma transição singlete -> singlete e por isso a emissão a partir de tal transição (T1 -> S0) pode persistir por algum tempo após a irradiação para excitação ter sido interrompida.
A conversão interna e externa competem com tamanha eficiência com a fosforescência que esse tipo de emissão é normalmente observado apenas a baixas temperaturas, em meios altamente viscosos ou em moléculas adsorvidas em superfícies sólidas.
Além disso, as bandas de fosforescência são encontradas geralmente em comprimentos de onda maiores que as bandas de fluorescência porque o estado triplete excitado tem, em muitos casos, energia menor que o estado singlete correspondente.
O processo de fluorescência é regido por três importantes eventos (excitação, relaxamento vibracional e emissão) que ocorrem em escalas de tempo que são separados por várias ordens de grandeza.
A excitação de uma molécula acontece em femtossegundos (10-15 segundos), o relaxamento vibracional dos elétrons pode ser medido em picossegundos (10-12 segundos) e a emissão de um fóton ocorre em um período de tempo relativamente longo de nanossegundos (10-9 segundo).
Tabela 8. Escala de tempo dos processos envolvidos na Fluorescência e Fosforescência (Fonte: www.olympusfluoview.com/theory/fluoroexciteemit.html
| Transição | Processo | Constante de velocidade | Tempo (s) |
|---|---|---|---|
| S0 -> S1 ou S2 ou Sn | Absorção (Excitação) | Instantânea | 10-15 |
| Sn -> S1 | Conversão Interna | kci | 10-14 - 10-10 |
| S1 -> S1 | Relaxação Vibracional | krv | 10-12 - 10-10 |
| S1 -> S0 | Fluorescência | kf | 10-9 - 10-7 |
| S1 -> T1 | Cruzamento Intersistema | ki | 10-10 - 10-8 |
| S1 -> S0 | Supressão (Conversão Externa) | ks (ou kq) | 10-7 - 10-5 |
| T1 -> S0 | Fosforescência | kp | 10-3 - 100 |
O Tempo de Vida (τ) do estado excitado é definido é definido como o tempo médio que uma molécula permanece no estado excitado antes de retornar ao estado fundamental.
O tempo de vida (τ) pode ser calculado pelo inverso do somatório das constantes de velocidade de todos os processos de desativação do estado excitado.
τ = 1 / ( kf + knr )
Onde knr representa o somatório das constantes de velocidade de todos os demais processos de desativação não-radiativos (kci, ki, ks ...)
A figura seguinte ilustra o decaimento da fluorescência em função do tempo segundo uma função exponencial:
It = I0 x e(-t/τ)
Onde:
It é a intensidade de fluorescência medida no tempo t
I0 é a intensidade de fluorescência inicial imediatamente após a excitação
τ é o tempo de vida de fluorescência
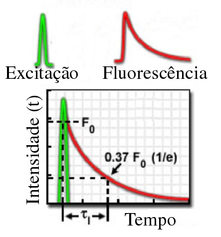
A emissão de fluorescência é um processo aleatório e poucas moléculas emitem seus fótons exatamente no tempo t = τ e portanto τ representa um tempo médio de permanência no estado excitado.
Em um decaimento monoexponencial, como ilustrado na figura anterior, 63% das moléculas decaem até o tempo t = τ, e 37% decaem no restante do tempo (t > τ).
As duas técnicas mais comuns para medida do tempo de vida da fluorescência são o método pulsado e o método de modulação de fase. (Fluorometria no domínio da frequência)
O rendimento quântico, ou eficiência quântica, para fluorescência ou fosforescência é simplesmente a razão do número de moléculas que emitem luz pelo número total de moléculas excitadas ou em outras palavras, o número de fótons emitidos em relação ao número de fótons absorvidos.
Com base no diagrama de Jablonski e considerando os processo de desativação já discutidos podemos definir o rendimento quântico de fluorescência Φ de um composto como a razão entre a constante de velocidade de fluorescência kf e o somatório das constantes de velocidade de todos os processos de desativação do estado excitado singlete mais baixo:
Φ = kf / ( kf + kci + ki + ks )
Quanto mais rápida for a desativação por fluorescência (alto kf) em relação aos processos não-radiativos mais intensa será a fluorescência observada.
E a recíproca é verdadeira, ou seja, se um processo não-radiativo tem uma constante de velocidade mais favorável, ou não há fluorescência ou é pouco intensa.
Para uma molécula altamente fluorescente, como a fluoresceína, o rendimento quântica em algumas condições se aproxima da unidade.
Figura 147. Molécula de Fluoresceína e os espectros de excitação (com medida da emissão em 520 nm) e emissão (com excitação em 480 nm)(Fonte: http://pt.wikipedia.org/wiki/Fluoresceína
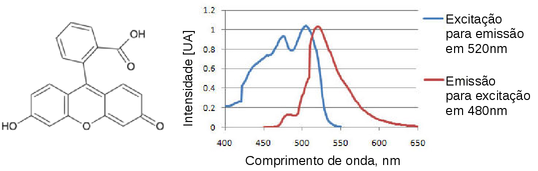
O espectro de excitação da figura acima é obtido medindo-se a entensidade de luminescência em um comprimento de onda fixo (normalmente no comprimento de onda de máxima emissão), enquanto o comprimento de onda de excitação é variado.
Como a primeira etapa para se gerar emissão de fluorescência é a absorção de radiação, um espectro de excitação é essencialmente idêntico ao espectro de absorção registrado nas mesmas condições.
Os espectros de emissão (Fluorescência ou Fosforescência), por outro lado, são obtidos fazendo a excitação em um comprimento de onda fixo (normalmente no comprimento de onda de máxima absorção), enquanto se registra a intensidade de emissão em função do comprimento de onda.
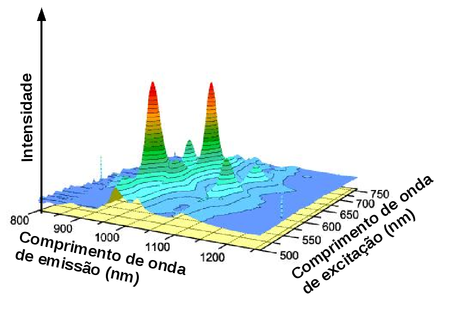
Portanto é possível obter o espectro total de excitação e emissão de um composto como um gráfico 3D onde um dos eixos corresponde aos comprimentos de onda de excitação, o segundo eixo corresponde aos comprimentos de onda de emissão e o terceiro eixo exibe a intensidade relativa de luminescência, conforme ilustrado na figura seguinte.
Figura 148. Gráfico de superfície 3D da intensidade de fotoluminescência de nanotubos de carbono de parede única em suspensão na água como uma função de excitação e emissão de comprimentos de onda. (Fonte: http://www.originlab.com)
Nota
A fluorescência dificilmente resulta da absorção de radiação ultravioleta de comprimentos de onda menores que 250 nm, porque a partir desse limite a radiação é suficiente energética para causar ruptura de ligações químicas e a desativação dos estados excitados.
É importante lembrar que o rendimento quântico não é um valor constante para uma molécula mas pode variar em função da polaridade do solvente, pH e concentração.
A tabela seguinte mostra valores de rendimento quântico para algumas substâncias fluorescentes.
Tabela 9. Rendimento quântico de fluorescência de algumas moléculas. (Fonte: http://micro.magnet.fsu.edu
| Substância | Solvente | Excitação (nm) | Emissão (nm) | Rendimento Quântico |
|---|---|---|---|---|
| Laranja de Acridina | Etanol | 493 | 535 | 0,46 |
| Benzeno | Etanol | 248 | 300-350 | 0,04 |
| Clorofila A | Etanol | 440 | 685 | 0,23 |
| Eosina Y | Água | 521 | 544 | 0,16 |
| Fluoresceína | Água | 537 | 515 | 0,92 |
| Rodamina B | Etanol | 555 | 627 | 0,97 |
Para muitas moléculas fluorescentes os espaçamentos dos níveis de energia vibracional no estado fundamental e excitado é muito semelhante o que resulta em um aspecto especular do espectro de emissão em relação ao espectro de absorção, ou seja, o espectro de emissão lembra a imagem especular do espectro de absorção referente à transição S0 -> S1.
No estado excitado as moléculas decaem rapidamente por relaxação vibracional para o menor nível vibracional do estado excitado e partir daí decaem por fluorescência para os níveis vibracionais superiores do estado fundamental sofrendo em seguida nova relaxação vibracional até o menor nível vibracional do estado fundamental.
Além disso a distribuição de probabilidades das transições de excitação são semelhantes à distribuição de probalidades das transições de emissão [12] contribuindo para a simetria entre os espectros de absorção e emissão.
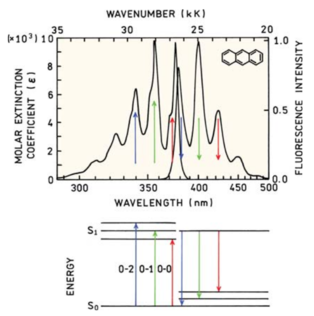
Figura 149. Diagrama esquemático de um espectro hipotético de absorção com bandas referentes a 3 transições vibrônicas S0 -> S1 (0,0; 0,1 e 0,2) e o respectivo espectro de emissão com 3 bandas vibrônicas S1 -> S0 com a superposição das bandas 0,0. (Fonte: Curso Prof. Amilcar Machulek Junior (IQ/USP))
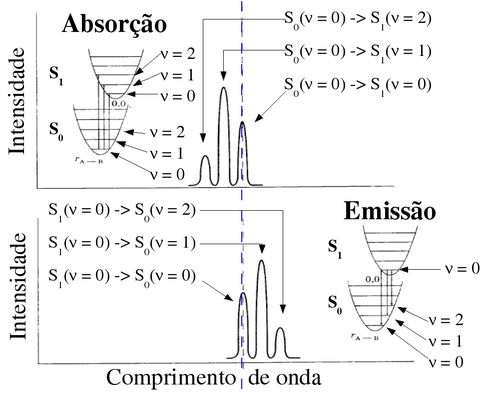
Na prática, o perfil do espectro da figura anterior não é usualmente obtido com tanta definição pois o pequeno espaçamento entre os níveis de energia vibracional do estado fundamental e excitado associada à agitação térmica dá origem a uma superposição dos picos das transições vibrônicas que acaba resultando, principalmente em fase condensada, em bandas de absorção e emissão largas e sem estrutura. (Fonte: Química Nova)
Figura 150. Exemplo do “regra da imagem-espelho” nos espectros de absorção e emissão da molécula de antraceno.(Fonte: Lakowicz, 2006)
Nota
Uma estratégia para melhor visualizar a regra da imagem-espelho é visualizar o espectro de absorção e emissão em função do “número de onda” (o inverso do comprimento de onda) o qual representa o número de ondas por unidade de comprimento e é diretamente proporcional à frequência (= energia).
Existem exceções a esta regra e o Quinino é um exemplo.
A fluorescência pode ser reduzida por uma série de mecanismos, dentre eles destacamos a Supressão como um processo reversível e a Fotodegradação como um processo irreversível.
Figura 151. Duas amostras de quinino dissolvido em água com um laser violeta (à esquerda) iluminando ambas. Pode-se observar a fluorescência azul do quinino no copo da direita. A amostra da esquerda contém íons cloreto, que suprime a fluorescência do quinino, e por isso observa-se apenas a luz violeta refratada do laser.(Fonte: http://en.wikipedia.org/)

A supressão da fluorescência pode ocorrer por diferentes mecanismos de relaxação não-radiativa do estado excitado para o estado fundamental, em escala intramolecular ou intermolecular.
Figura 152. Decaimento radiativo e não-radiativo. (kf é a constante de velocidade de fluorescência e knr representa o somatório das constantes de velocidade de todos os demais processos de desativação não-radiativos)
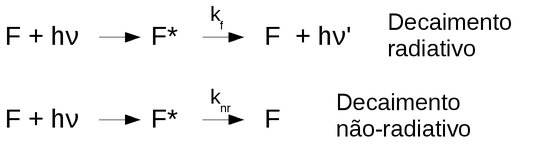
A supressão colisional (ou dinâmica) intermolecular ocorre quando a molécula no estado excitado sofre um decaimento não-radiativo pelo contato com alguma outra molécula (supressor), sem reação química entre ambas.
Na supressão dinâmica a queda da intensidade de fluorescência é descrita pela equação de Stern Volmer.
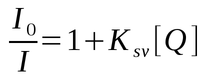
Onde Ksv é a constante Stern-Volmer, I e I0 representam respectivamente a intensidade de emissão na presença do supressor e a intensidade de emissão na ausência do supressor, e [Q] é a concentração do supressor.
Figura 154. Espectros de fluorescência com quantidades crescentes de um supressor, equação de Stern-Volmer e o respectivo gráfico (I0/I em função de [Q]) indicando Ksv como a inclinação da reta. (Fonte: Journal of Chinese Clinical Medicine;2008,2;Vol.3,No.2)
![Espectros de fluorescência com quantidades crescentes de um supressor, equação de Stern-Volmer e o respectivo gráfico (I0/I em função de [Q]) indicando Ksv como a inclinação da reta. (Fonte: Journal of Chinese Clinical Medicine;2008,2;Vol.3,No.2)](figuras//grafico_stern_volmer.png)
A constante de Stern-Volmer (Ksv) indica a sensibilidade e/ou acessibilidade do fluoróforo ao supressor.
Por exemplo, um fluoróforo (sonda fluorescente) disperso em uma matriz polimérica está geralmente inacessível a um supressor solúvel em água, e neste caso (Ksv) é pequeno. Grandes valores de Ksv são obtidos quando o fluoróforo está livre em solução ou na superfície de uma biomolécula.
A constante de Stern-Volmer também pode ser definida como o produto Ksv = kq x τ0, onde kq é a constante de supressão bimolecular (M-1 . s-1) e τ0 é o tempo de vida na ausência do supressor.
Nota
A supressão colisional intramolecular pode ocorrer quando o fluoróforo e supressor estão interligados por ligações covalentes e portanto fazem parte da mesma molécula. Uma técnica utilizada no estudo da dinâmica do DNA.
Na supressão dinâmica com decaimento monoexponencial a redução na intensidade de fluorescência é equivalente à redução no tempo de vida
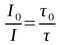 e portanto a equação de Stern-Volmer pode ser descrita em função dos tempos de vida:
e portanto a equação de Stern-Volmer pode ser descrita em função dos tempos de vida:
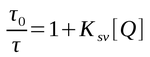
onde τ0 é o tempo de vida na ausência do supressor e τ é o tempo de vida na presença do supressor.
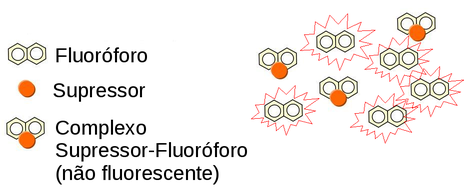
Na supressão estática o fluoróforo e o supressor formam um complexo estável não fluorescente, o qual absorve luz mas retorna ao estado fundamental sem emissão de fóton.
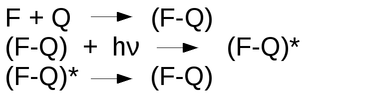
Equação 2. Dependência da fluorescência em função da concentração do supressor em um mecanismo de supressão estática.
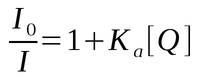
No caso da supressão estática Ka representa a constante de associação do complexo:
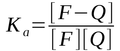
Figura 156. Na supressão estática apenas os fluoróforos complexados deixam de fluorescer. (Fonte: www.physics.umn.edu)
A supressão estática não altera as propriedades dos fluoróforos não complexados, os quais continuam ativos para fluorescer, mas apenas reduz o número de moléculas fluorescentes ativas e portanto o tempo de vida do estado excitado não é alterado (τ). Por isso a redução da intensidade de fluorescência não é equivalente ao tempo de vida:
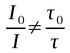 .
.
Os gráficos de I0/I e τ0/τ em função de [Q] assumem diferentes perfis conforme e mecanismo de supressão, conforme ilustra a figura seguinte.
Figura 157. Gráficos de I0/I e τ0/τ em função de [Q] para diferentes mecanismos de supressão. Gráfico 1 - supressão dinâmica. Gráfico 2 - supressão estática. Gráfico 3 - combinação de supressão estática e dinâmica. (Fonte: http://commons.wikimedia.org e http://de.wikipedia.org)
![Gráficos de I0/I e τ0/τ em função de [Q] para diferentes mecanismos de supressão. Gráfico 1 - supressão dinâmica. Gráfico 2 - supressão estática. Gráfico 3 - combinação de supressão estática e dinâmica. (Fonte: http://commons.wikimedia.org e http://de.wikipedia.org)](figuras/supr_est_dinam_comb.png)
Quando ambos os mecanismos (dinâmico e estático) estão atuando no sistema as equações de supressão dinâmica e de supressão estática são combinadas, resultando na equação:
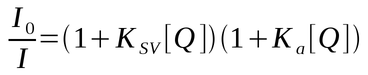
O desvio positivo da linearidade que aparece no Gráfico 3 é devido ao termo quadrático [Q]2 que aparece no desenvolvimento da equação combinada acima.
Portanto além da medida de intensidade de fluorescência, a medida do tempo de vida de fluorescência é um parâmetro importante para esclarecer se o mecanismo de supressão predominante é estático, dinâmico ou a combinação de ambos.
Em alguns casos é utilizado um modelo fenomenológico (ou empírico) [13] para descrever a supressão estática:
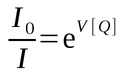
onde V representa o elemento de volume ativo em torno do fluoróforo e dentro do qual um supressor pode suprimir o fluoróforo excitado. (Fonte: www.fluorescence-foundation.org)
Quando ambos os mecanismos (dinâmico e estático) estão atuando no sistema, as equações de supressão dinâmica e de supressão estática (empírica) são combinadas, resultando na equação:
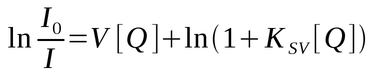
Em alguns sistemas, a curva de supressão desvia da linearidade tendendo a um patamar sugerindo a presença de uma fração do fluoróforo não acessível ao supressor, conforme a figura seguinte.
Figura 158. Gráfico de Stern-Volmer não linear sugerindo a presença de uma fração do fluoróforo não acessível ao supressor. (Fonte: Nei M.C.G., 1997)
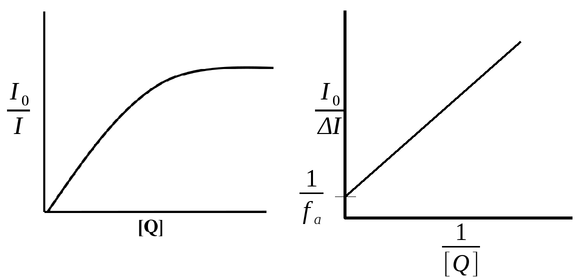
A fração acessível (fa) e a constante de Stern-Volmer para este caso pode ser calculada pela forma modificada da equação de Stern-Volmer:
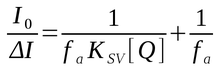
onde ΔI = (I0 - I) e fa = I0a/(I0i + I0a) é a fração da fluorescência inicial acessível ao supressor e KSV é a constante de Stern-Volmer da fração acessível, I0a é a intensidade de fluorescência inicial acessível ao supressor e I0i é a fração de fluorescência inicial inacessível ao supressor. (Nei M.C.G., 1997)
Esse modelo assume a existência de duas populações de fluoróforo, uma acessível e outra inacessível ao supressor, mas ambas com os mesmos parâmetros de fluorescência.
Portanto quando o gráfico I0/ΔI em função de 1/[Q] é linear, a interseção permite estimar o parâmetro e fa e a partir da inclinação pode-se obter o KSV.
Esse modelo foi usado para tratar os dados de supressão de fluorescência de resíduos de triptofano em proteínas.(Photochem Photobiol, 2000)
Vimos até aqui reações entre fluoróforo e supressor que não modificam a estrutura química de ambos e portanto são processos físicos.
No entanto uma molécula fluorescente pode também sofrer reações químicas no estado excitado devido a mudanças na distribuição eletrônica do fluoróforo no estado excitado.
O fenol é um exemplo de fluoróforo que aumenta a acidez no estado excitado.
Os fluoróforos, no estado excitado, também pode sofrer reações irreversíveis de Fotodegradação perdendo a sua capacidade de fluorescer.
A maioria dos fluoróforos pode repetir ciclos de excitação e emissão centenas ou milhares de vezes antes de sofrer fotodegradação, resultando na perda de fluorescência. Por exemplo, o Isotiocianato de Fluoresceína (FITC) pode sofrer aproximadamente 30.000 ciclos de excitação e decaimento antes de sofrer fotodegradação.
Alguns fluoróforos se degradam rapidamente após emitir apenas alguns fótons, enquanto outros podem sofrer milhares ou milhões de ciclos antes de se degradar.
Uma molécula no estado excitado também pode transferir sua energia eletrônica para outro molécula sem contato direto através da Transferência de Energia Ressonante, ou por Ressonância, (RET - Resonance Energy Transfer).
A RET é um processo que não envolve a emissão e reabsorção de fótons, ou seja, não é o resultado da emissão de um fóton de uma molécula “Doadora (D)” sendo absorvido por uma molécula “Aceptora (A)” (fluorescente ou não).
O mecanismo da RET baseia-se no conceito do fluoróforo como um dipolo oscilante, o qual pode trocar energia com outro dipolo que possua uma frequência de ressonância similar, similar aos osciladores acoplados. (Fonte: Fabrício C.L.L., 2009).
A Transferência de Energia por Ressonância depende da distância entre “D” e “A” e da sobreposição dos espectros de emissão de “D” e de absorção de “A”
A presença de oxigênio dissolvido frequentemente reduz a intensidade de fluorescência de uma solução. Esse efeito pode ser o resultado de uma oxidação da espécie fluorescente, induzida fotoquimicamente.
Mais comumente, no entanto, a supressão acontece como consequência das propriedades paramagnéticas do oxigênio molecular, que promove cruzamento intersistema e conversão das moléculas excitadas ao estado triplete.
Outras espécies paramagnéticas também tendem a suprimir a fluorescência.
Nota
Nesta seção transcrevemos literalmente muitas informações disponíveis na Tese de Doutoramento: “Desenvolvimento de sensores de Oxigênio Dissolvido utilizando métodos eletroquímicos e ópticos para monitoramento em tempo real da qualidade da água” (Ferreira M.A.C., 2007)
Os sensores ópticos para OD, também chamados de optodos ou optrodos, se baseiam na capacidade do Oxigênio de suprimir a Fluorescência emitida por um Fluoróforo. E portanto a intensidade do efeito de supressão da fluorescência pode ser correlacionada com a concentração do Oxigênio.
As vantagens destes sensores são basicamente: nenhum consumo de oxigênio no processo, não requer nenhum eletrodo de referência, insensível a taxas de fluxo ou velocidade da amostra, imune ao campo elétrico exterior, precisão similar ao método de titulação Winkler e baixa sensibilidade à presença de H2S. Também são imunes a interferentes químicos tais como Cl2, SO2 e H2O2 em baixas concentrações. Uma limitação, porém desses sensores é a forte influência da temperatura no processo de redução da luminescência.
Os sensores de oxigênio ópticos são constituídos basicamente por um indicador luminescente e por um sistema óptico. O indicador luminescente sensível ao oxigênio pode estar impregnado em uma membrana ou um sol-gel. O sistema óptico inclui a fonte de excitação (LEDs ou laser), filtros ópticos para seleção do comprimento desejado, guias de onda (fibra ótica), fotodetectores como fotodiodo ou tubo fotomultiplicador, além do sistema de controle e processamento do sinal.
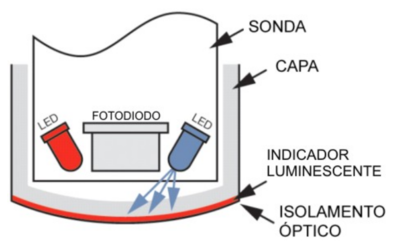
O sensor da figura anterior utiliza um par de LEDs (azul e vermelho) e um fotodetector de silício. A capa sensora contém um indicador luminescente (platina) excitado pelo LED azul. O indicador é protegido do ambiente externo com uma camada de isolamento óptico preto (poliestireno carbonato), evitando a interferência de fontes externas de luz quando a capa sensora esta acoplada à sonda. O circuito de medição para este sensor está baseado no tempo de vida da luminescência, determinado através de uma diferença de fase entre a modulação senoidal do LED azul e a luminescência detectada pelo fotodiodo.
Os fluoróforos mais comumente empregados na determinação de oxigênio dissolvido são os hidrocarbonetos policíclicos aromáticos (pirenos e seus derivados, quinolina e fenantreno), complexos de metais de transição (Ru+2, Os+2, Ir+2) e metaloporfirinas (Pt+2, Pd+2, Zn+2). (Silva K.R.B., 2007)
Os complexos de Rutênio II são os mais empregados devido a sua acentuada absorção na faixa visível do espectro, seu alto rendimento quântico, maior tempo de vida do estado excitado e a boa distinção entre os comprimentos de onda de excitação e emissão.
Os fluoróforos podem ser imobilizados em diversos suportes, tais como: filme sol-gel de sílica porosa, polímero acrílico, borracha de silicone, poliestireno e outros. São desejáveis os polímeros permeáveis ao oxigênio, solubilidade para incorporação do fluoróforo e elevada estabilidade em relação à fotodegradação.
As vantagens do uso do processo sol-gel na imobilização de espécies incluem:
condições de processamento à temperatura ambiente;
porosidade ajustável do filme
boa estabilidade térmica
transparência óptica
procedimentos simples para incorporação dos dopantes
A maioria dos trabalhos publicados sobre sensores ópticos de OD é baseada na medida da supressão da fluorescência, porém esta técnica de transdutância apresenta alguns inconvenientes para sua mensuração. Os principais são a suscetibilidade à fonte de luz externa, movimento do sensor, alterações no caminho óptico, degradação ou lixiviação do indicador. Estes efeitos podem ser minimizados pelo uso do tempo de vida ao invés da intensidade de fluorescência.
A medida do tempo de vida da fluorescência mostra algumas vantagens importantes sobre a medida de intensidade, porque o tempo de decaimento não é influenciado pela concentração do indicador, sua deterioração, variação na fonte de excitação ou sensibilidade do fotodetector, variações no caminho óptico ou a espessura do filme. Assim, não é necessário corrigir a luz de emissão em função da luz da excitação, e o sistema é insensível à sujeira em elementos ópticos.
Nota
As principais diferenças entre os métodos de medida é que a intensidade de luminescência é uma grandeza extensiva enquanto a medida do tempo de vida é uma grandeza intensiva.
A quantificação extensiva (volume ou energia) depende do tamanho da amostra, enquanto que uma quantificação intensiva (temperatura ou concentração) é independente do tamanho da amostra.
Os sensores ópticos de OD geralmente são calibrados por um dos diversos modelos de aproximação baseados na equação linear de Stern-Volmer.
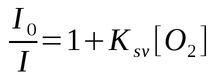
Quanto maior o KSV menor o limite de detecção e portanto maior a sensibilidade para o sistema de detecção.
Um fluoróforo incorporado em uma matriz polimérica não segue a equação de Stern-Volmer original e por isso vários modelos matemáticos são propostos para a calibração dos sensores.
O modelo mais citado é baseado no conceito de micro-ambientes heterogêneos:
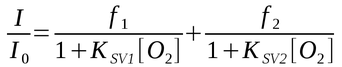
onde KSV1 e KSV2 são constantes de Stern-Volmer para os dois micro-ambientes e f1 e f2 são suas frações de contribuição.
Embora sistemas de membranas sejam na realidade múltiplos ambientes, o modelo duplo é suficiente para a maioria das aplicações de calibração.
Uma modificação da equação Stern-Volmer exclui a necessidade de fornecimento da intensidade para o nível zero de oxigênio (I0) para a equação de calibração:
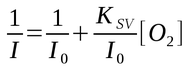
A partir do gráfico de 1/I em função de [O2] é possível obter I0 a partir da interseção e KSV é obtido pela inclinação.
O aumento na temperatura reduz a intensidade da fluorescência, com maior ou menor intensidade dependendo do composto.
O efeito da temperatura no aumento da supressão está relacionado com o aumento da agitação molecular e consequente aumento das colisões entre as moléculas.
Portanto equações para a calibração de resposta do sensor óptico devem levar em consideração a variação de temperatura.
A seguir alguns links para apresentações muito informativas sobre fluorescência.
Basic Spectroscopic Principles - Joachim Mueller.
Fluorescence Principles - Joachim Mueller
Steady-state Fluorometer - Joachim Mueller
Quantum Yield and Polarization (1) - Joachim Mueller
Fluorescence Lifetime + Polarization (2) - Joachim Mueller
Quenching and FRET - Joachim Mueller
Introduction to Fluorescence Essays - Joachim Mueller
Fluorescence Microscopy and Fluorescence Correlation Spectroscopy - Joachim Mueller
[6] Polarografia é um tipo particular de Voltametria que foi descoberto pelo químico tchecoslovaco Jaroslav Heyrovsky no início dos anos 1920. A Polarografia difere de outros tipos de Voltametria porque o microeletrodo tem a forma de um eletrodo gotejante de mercúrio. Portanto o termo Polarografia não é adequado para esses eletrodos mas se tornou comum na literatura.Skoog, 2002
[7] O comprimento de ligação pode ser determinado por difração de raios X em sólidos, por difração de elétrons e por métodos espectroscópicos (estudo da luz absorvida ou emitida por moléculas). (Fonte: www.science.uwaterloo.ca
[8] Faixa do Visível (Vis) (380-400 nm < λ < 700-800 nm)
Faixa do Ultravioleta (UV) (200 < λ < 380-400 nm)
[9] O Spin é considerado hoje uma entidade matemática. O Spin não possui uma interpretação clássica, ou seja, é um fenômeno estritamente quântico, e sua associação com o movimento de rotação das partículas sobre seu eixo - uma visão clássica - é apenas um recurso didático que deixa muito a desejar.(Wikipedia)
[10] Vapores Atômicos Diluídos são utilizados na técnica analítica de Espectroscopia de Absorção Atômica na qual o processo de Atomização pode ser feito em uma chama ou por via eletrotérmica.
Na Atomização por chama uma solução da amostra é nebulizada por um fluxo de oxidante gasoso, misturada com um combustível gasoso, e introduzida no interior de uma chama onde ocorre a atomização após uma sucessão de processos inter-relacionados. (Vídeo sobre a Espectroscopia de Absorção Atômica em Chama)
[11] Nascido na Ucrânia em 1898, Alexander Jablonski é mais conhecido como o pai da espectroscopia de fluorescência. O principal interesse científico de Jablonski foi a polarização da fotoluminescência em soluções. Seu trabalho é muito conhecido pelo Diagrama de Energia de Jablonski, uma ferramenta que pode ser usada para explicar a cinética e espectros de fluorescência, fosforescência e fluorescência atrasada. (Fonte: http://micro.magnet.fsu.edu/optics/timeline/people/jablonski.html
[12] Os espectros de excitação e de emissão podem ser consideradas como funções de distribuição de probabilidade de que um fóton de determinado quantum de energia seja absorvido e emitido respectivamente.
[13] Um modelo fenomenológico, ou empírico, expressa matematicamente os resultados dos fenômenos observados, sem ser deduzido a partir de uma teoria.
Expressões algébricas simples podem ser usadas para modelar as observações ou resultados experimentais. O modelo algébrico é então usado para fazer previsões sobre os resultados de outras observações ou experimentos. Se as previsões feitas pelo modelo algébrico são suficientemente precisos, muitas vezes são adotados pela comunidade científica, apesar do fato de que as expressões algébricas não tenham sido derivadas da teoria fundamental desse domínio de conhecimento. (Fonte: Phenomenology (science)