Nota
As principais fontes de informação para essa seção foram: Introduction to multivariate calibration in analytical chemistry, 2000 e Introduction to Multivariate Calibration - A Practical Approach, 2018.
A seção Hackeando as Águas tem como um dos objetivos o desenvolvimento de sistemas para a análise de águas. E uma das etapas fundamentais para o uso de analisadores é a “calibração”.
Uma definição de calibração:
Calibração (ou aferição) Conjunto de operações que estabelece, sob condições especificadas, a relação entre os valores indicados por um instrumento de medição ou sistema de medição ... , e os valores correspondentes das grandezas estabelecidas por padrões.(Fonte: glossario-de-metrologia.pdf)
Essa relação é definida por uma equação matemática que estabelece uma relação entre uma variável (univariado) ou várias variáveis (multivariado) independentes (medidas) e uma ou mais grandezas dependentes (respostas).
Quando usamos apenas uma variável independente temos uma “Calibração Univariada”. E quando utilizamos mais de uma variável independente para a determinação da grandeza de interesse, temos uma “Calibração Multivariada”.
Mas uma “Equação de Calibração” é um tipo específico de “Modelo” (neste caso um “Modelo Matemático”).
Como foi visto na seção sobre (Regressão Linear Univariada) a calibração Univariada busca uma equação matemática que estabelece uma relação entre uma variável “independente” (x) e uma grandeza “dependente” (y), as quais podem ser organizadas em uma tabela com uma coluna para x (variável independente) e outra coluna para y (variável dependente).
Alguns exemplos de calibração univariada: Gráfico ORP x -log[Cl2], Gráficos da relação de Transmitância e Absorbância para diferentes concentrações de permanganato de potássio e Gráfico de calibração de uma balança.
Vamos exemplificar o uso de diferentes estratégias de calibração “univariada” usando como base de dados (dataset) um conjunto de 25 espectros na região do UV (220-350 nm) de 25 soluções formadas pela mistura de 10 Hidrocarbonetos Poliaromáticos (HPA):
Py - pyrene;
Ace - acenaphthene;
Anth - anthracene;
Acy - acenaphthylene;
Chry - chrysene;
Benz - benzanthracene;
Fluora - fluoranthene;
Fluore - fluorene;
Nap - naphthalene;
Phen - phenanthrene.
Vamos usar nesse exemplo o mesmo procedimento que foi explicado na seção Modelos Empíricos com Regressão Linear Univariada mas utilizando o seguinte conjunto de dados:
25 espectros de absorção eletrônica na região do UV (220-350 nm),
em 27 comprimentos de onda na faixa de 220-350nm com intervalos de 5 nm,
de 25 soluções formadas pela mistura de 10 Hidrocarbonetos Poliaromáticos (HPA),
onde cada amostra é uma mistura dos 10 HPA em 5 diferentes concentrações.
Os 25 espectros estão disponíveis para download no link tabela_25_espectros_UV_Vis_25_comp_onda_220_350_de_mist_10_PAH.csv.
E a tabela com as 25 amostras com 5 diferentes concentrações dos 10 Hidrocarbonetos Poliaromáticos está disponível para download no link tabela_25_solucoes_10_PAH.csv.
Na calibração univariada clássica o objetivo é determinar a concentração de um único analito usando a resposta de um único “detector”, por exemplo a intensidade de absorção ou emissão em um único comprimento de onda, ou a área de um único pico cromatográfico.
Para isso, uma série de medidas são realizadas para correlacionar a medida espectroscópica (ou outro sinal analítico) à concentração conforme a relação:
Y ≈ C × B
Onde “y” é um vetor coluna formado pelas respostas do sensor (Ex: absorbância em um comprimento de onda) para um conjunto de amostras que formam o vetor “c” que contêm as concentrações do analito de interesse em diferetes concentrações. E “b” é um “escalar” que correlaciona os dois vetores (y e c) e é determinado pela técnica de Regressão.
Ambos os vetores (Y e C) possuem o mesmo número de linhas correspondente ao número de amostras.
O escalar “b” é determinado pela equação U.1.
Equação U.1. Equação matricial geral para o ajuste de modelos pela técnica dos “mínimos quadrados” para a “calibração univariada clássica”.
B ≈ (CtC)-1 CtY
Nota
Note que se trata da mesma equação matricial geral para regressão por mínimos quadrados, mas agora “y” representa os sinais gerados nas diferentes concentrações (c) do analito de interesse.
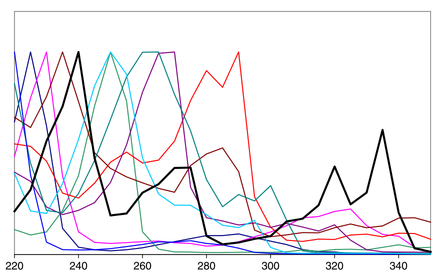
A figura U.1 mostra os espectros de UV (220-350nm) de cada um dos 10 Hidrocarbonetos Poliaromáticos sobrepostos. E pode-se constatar que o Pireno (Py) é praticamente a única substância que apresenta uma absorbância significativa em 335 nm.
Figura U.1. Espectros de aborbância sobrepostos, na região de 220 a 350 nm, de cada um dos PAH. O espectro do pireno (Py) aparece destacado em preto com uma absorbância significativa em 335nm. (Fonte: Chemometrics - Data Driven Extraction for Science, 2018
Por isso vamos visualizar os dados do pireno e usar os comandos do Python para colocar em um gráfico os valores de absorbância no comprimento de onda de 335 nm para as diferentes concentrações nas 25 soluções de mistura..
A tabela com a composição de 25 amostras contendo misturas dos 10 Hidrocarbonetos Poliaromáticos em diferentes concentrações (disponível para download no link tabela_25_solucoes_10_PAH.csv), apresenta a seguinte estrutura:
,Polyarene conc,,mg L-1,,,,,,, Spectrum,Py,Ace,Anth,Acy,Chry,Benz,Fluora,Fluore,Nap,Phen 1,"0,456","0,120","0,168","0,120","0,336","1,620","0,120","0,600","0,120","0,564" 2,"0,456","0,040","0,280","0,200","0,448","2,700","0,120","0,400","0,160","0,752" 3,"0,152","0,200","0,280","0,160","0,560","1,620","0,080","0,800","0,160","0,118" 4,"0,760","0,200","0,224","0,200","0,336","1,080","0,160","0,800","0,040","0,752" 5,"0,760","0,160","0,280","0,120","0,224","2,160","0,160","0,200","0,160","0,564" 6,"0,608","0,200","0,168","0,080","0,448","2,160","0,040","0,800","0,120","0,940" 7,"0,760","0,120","0,112","0,160","0,448","0,540","0,160","0,600","0,200","0,118" 8,"0,456","0,080","0,224","0,160","0,112","2,160","0,120","1,000","0,040","0,118" 9,"0,304","0,160","0,224","0,040","0,448","1,620","0,200","0,200","0,040","0,376" 10,"0,608","0,160","0,056","0,160","0,336","2,700","0,040","0,200","0,080","0,118" 11,"0,608","0,040","0,224","0,120","0,560","0,540","0,040","0,400","0,040","0,564" 12,"0,152","0,160","0,168","0,200","0,112","0,540","0,080","0,200","0,120","0,752" 13,"0,608","0,120","0,280","0,040","0,112","1,080","0,040","0,600","0,160","0,376" 14,"0,456","0,200","0,056","0,040","0,224","0,540","0,120","0,800","0,080","0,376" 15,"0,760","0,040","0,056","0,080","0,112","1,620","0,160","0,400","0,080","0,940" 16,"0,152","0,040","0,112","0,040","0,336","2,160","0,080","0,400","0,200","0,376" 17,"0,152","0,080","0,056","0,120","0,448","1,080","0,080","1,000","0,080","0,564" 18,"0,304","0,040","0,168","0,160","0,224","1,080","0,200","0,400","0,120","0,118" 19,"0,152","0,120","0,224","0,080","0,224","2,700","0,080","0,600","0,040","0,940" 20,"0,456","0,160","0,112","0,080","0,560","1,080","0,120","0,200","0,200","0,940" 21,"0,608","0,080","0,112","0,200","0,224","1,620","0,040","1,000","0,200","0,752" 22,"0,304","0,080","0,280","0,080","0,336","0,540","0,200","1,000","0,160","0,940" 23,"0,304","0,200","0,112","0,120","0,112","2,700","0,200","0,800","0,200","0,564" 24,"0,760","0,080","0,168","0,040","0,560","2,700","0,160","1,000","0,120","0,376" 25,"0,304","0,120","0,056","0,200","0,560","2,160","0,200","0,600","0,080","0,752"
A primeira linha (,Polyarene conc,,mg L-1,,,,,,,) traz informações sobre a unidade de concentração dos PAH (mg/L-1), mas vamos descartar quando ela for importada pelo comando read_csv do Pandas, usando o parâmetro skiprows = 1
Primeiro carregar as bibliotecas:
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
#Para usar toda a largura do terminal
pd.set_option('display.width', pd.util.terminal.get_terminal_size()[0])
Depois ler o arquivo csv e armazenar na variável data_solucoes:
data_solucoes = pd.read_csv ('tabela_25_solucoes_10_PAH.csv', sep = ',', skiprows = 1, decimal=b',', skipinitialspace=True)
print(data_solucoes)
No arquivo csv original a vírgula estava sendo usada como separador de campo e como separador decimal, por isso foi especificado os parâmetros sep = ',' e decimal=b',' no comando read_csv. E o parâmetro skiprows = 1 especifica que deve ser ignorada a primeira linha que contém informações sobre a unidade de concentração (,Polyarene conc,,mg L-1,,,,,,,).
O comando print(data_solucoes) indica que que os dados foram importados corretamente e a vírgula (como separador decimal) foi substituída por ponto:
Spectrum Py Ace Anth Acy Chry Benz Fluora Fluore Nap Phen 0 1 0.456 0.12 0.168 0.12 0.336 1.62 0.12 0.6 0.12 0.564 1 2 0.456 0.04 0.280 0.20 0.448 2.70 0.12 0.4 0.16 0.752 2 3 0.152 0.20 0.280 0.16 0.560 1.62 0.08 0.8 0.16 0.118 3 4 0.760 0.20 0.224 0.20 0.336 1.08 0.16 0.8 0.04 0.752 4 5 0.760 0.16 0.280 0.12 0.224 2.16 0.16 0.2 0.16 0.564 5 6 0.608 0.20 0.168 0.08 0.448 2.16 0.04 0.8 0.12 0.940 6 7 0.760 0.12 0.112 0.16 0.448 0.54 0.16 0.6 0.20 0.118 7 8 0.456 0.08 0.224 0.16 0.112 2.16 0.12 1.0 0.04 0.118 8 9 0.304 0.16 0.224 0.04 0.448 1.62 0.20 0.2 0.04 0.376 9 10 0.608 0.16 0.056 0.16 0.336 2.70 0.04 0.2 0.08 0.118 10 11 0.608 0.04 0.224 0.12 0.560 0.54 0.04 0.4 0.04 0.564 11 12 0.152 0.16 0.168 0.20 0.112 0.54 0.08 0.2 0.12 0.752 12 13 0.608 0.12 0.280 0.04 0.112 1.08 0.04 0.6 0.16 0.376 13 14 0.456 0.20 0.056 0.04 0.224 0.54 0.12 0.8 0.08 0.376 14 15 0.760 0.04 0.056 0.08 0.112 1.62 0.16 0.4 0.08 0.940 15 16 0.152 0.04 0.112 0.04 0.336 2.16 0.08 0.4 0.20 0.376 16 17 0.152 0.08 0.056 0.12 0.448 1.08 0.08 1.0 0.08 0.564 17 18 0.304 0.04 0.168 0.16 0.224 1.08 0.20 0.4 0.12 0.118 18 19 0.152 0.12 0.224 0.08 0.224 2.70 0.08 0.6 0.04 0.940 19 20 0.456 0.16 0.112 0.08 0.560 1.08 0.12 0.2 0.20 0.940 20 21 0.608 0.08 0.112 0.20 0.224 1.62 0.04 1.0 0.20 0.752 21 22 0.304 0.08 0.280 0.08 0.336 0.54 0.20 1.0 0.16 0.940 22 23 0.304 0.20 0.112 0.12 0.112 2.70 0.20 0.8 0.20 0.564 23 24 0.760 0.08 0.168 0.04 0.560 2.70 0.16 1.0 0.12 0.376 24 25 0.304 0.12 0.056 0.20 0.560 2.16 0.20 0.6 0.08 0.752
Vamos selecionar a coluna referente às concentrações de pireno (Py) na variável data_conc_py com os comandos:
#Seleciona a coluna referente ao pireno (Py)
data_conc_py = data_solucoes['Py']
#Converte do formato dataframe para array unidimensional
array_conc_py = data_conc_py.values[:]
#Converte para uma matriz coluna (bidimensional)
array_conc_py = array_conc_py.reshape(-1,1)
print("Concentração de pireno nas soluções: \n", array_conc_py)
E temos a saída:
Concentração de pireno nas soluções: [[0.456] [0.456] [0.152] [0.76 ] [0.76 ] [0.608] [0.76 ] [0.456] [0.304] [0.608] [0.608] [0.152] [0.608] [0.456] [0.76 ] [0.152] [0.152] [0.304] [0.152] [0.456] [0.608] [0.304] [0.304] [0.76 ] [0.304]]
O arquivo csv contendo os espectros apresenta a seguinte estrutura:
nm 220 225 230 235 240 245 250 255 260 265 270 275 280 285 290 295 300 305 310 315 320 325 330 335 340 345 350 1 0.771 0.714 0.658 0.537 0.587 0.671 0.768 0.837 0.673 0.678 0.741 0.755 0.682 0.633 0.706 0.290 0.208 0.161 0.135 0.137 0.162 0.130 0.127 0.165 0.110 0.075 0.053 2 0.951 0.826 0.737 0.638 0.738 0.911 1.121 1.162 0.869 0.870 0.965 1.050 0.993 0.934 1.008 0.405 0.254 0.185 0.157 0.159 0.180 0.155 0.150 0.178 0.140 0.105 0.077 3 0.912 0.847 0.689 0.514 0.504 0.622 0.805 0.892 0.697 0.728 0.790 0.728 0.692 0.639 0.717 0.292 0.224 0.168 0.134 0.123 0.136 0.115 0.095 0.102 0.089 0.068 0.048 4 0.688 0.679 0.662 0.558 0.655 0.738 0.838 0.883 0.670 0.656 0.704 0.668 0.562 0.516 0.573 0.295 0.212 0.172 0.138 0.144 0.179 0.130 0.134 0.191 0.107 0.060 0.046 5 0.873 0.801 0.732 0.640 0.750 0.820 0.907 0.955 0.692 0.681 0.775 0.866 0.798 0.749 0.827 0.311 0.213 0.170 0.151 0.162 0.201 0.158 0.170 0.239 0.146 0.094 0.067 6 0.953 0.850 0.732 0.612 0.724 0.882 1.013 1.087 0.860 0.859 0.925 0.938 0.869 0.810 0.868 0.390 0.241 0.175 0.138 0.141 0.167 0.129 0.135 0.178 0.115 0.078 0.056 7 0.613 0.577 0.547 0.448 0.508 0.463 0.473 0.549 0.510 0.560 0.604 0.493 0.369 0.343 0.363 0.215 0.178 0.165 0.136 0.142 0.183 0.122 0.129 0.193 0.089 0.041 0.030 8 0.927 0.866 0.799 0.605 0.618 0.671 0.771 0.842 0.646 0.658 0.716 0.829 0.783 0.731 0.794 0.292 0.220 0.155 0.120 0.123 0.144 0.122 0.127 0.164 0.113 0.078 0.056 9 0.585 0.577 0.543 0.450 0.515 0.614 0.734 0.815 0.624 0.642 0.724 0.716 0.680 0.649 0.713 0.298 0.213 0.167 0.136 0.126 0.137 0.109 0.104 0.129 0.098 0.074 0.057 10 0.835 0.866 0.803 0.550 0.563 0.577 0.642 0.732 0.675 0.713 0.836 0.938 0.911 0.847 0.910 0.358 0.226 0.173 0.153 0.160 0.186 0.156 0.157 0.193 0.134 0.093 0.066 11 0.477 0.454 0.450 0.433 0.538 0.593 0.661 0.724 0.554 0.561 0.616 0.469 0.355 0.322 0.345 0.211 0.154 0.139 0.114 0.118 0.154 0.100 0.100 0.154 0.071 0.030 0.016 12 0.496 0.450 0.402 0.331 0.389 0.504 0.613 0.599 0.383 0.344 0.353 0.366 0.338 0.309 0.334 0.194 0.123 0.094 0.076 0.070 0.077 0.065 0.056 0.065 0.053 0.036 0.025 13 0.594 0.512 0.441 0.392 0.489 0.562 0.637 0.647 0.415 0.387 0.421 0.461 0.396 0.361 0.424 0.161 0.103 0.076 0.062 0.076 0.110 0.082 0.094 0.144 0.078 0.043 0.028 14 0.512 0.478 0.409 0.319 0.375 0.395 0.436 0.487 0.439 0.449 0.474 0.438 0.362 0.331 0.360 0.204 0.158 0.122 0.089 0.087 0.107 0.075 0.079 0.114 0.064 0.040 0.028 15 0.662 0.583 0.586 0.564 0.687 0.708 0.748 0.757 0.611 0.581 0.641 0.727 0.638 0.596 0.658 0.277 0.171 0.133 0.113 0.128 0.168 0.125 0.143 0.211 0.114 0.067 0.044 16 0.768 0.588 0.501 0.386 0.429 0.539 0.671 0.740 0.607 0.627 0.693 0.772 0.751 0.710 0.781 0.287 0.174 0.116 0.093 0.089 0.095 0.087 0.081 0.087 0.081 0.069 0.047 17 0.635 0.557 0.487 0.377 0.404 0.488 0.597 0.669 0.603 0.625 0.647 0.576 0.515 0.477 0.524 0.260 0.188 0.139 0.105 0.096 0.104 0.084 0.071 0.077 0.061 0.045 0.031 18 0.575 0.489 0.432 0.375 0.408 0.449 0.525 0.569 0.442 0.451 0.501 0.519 0.474 0.458 0.500 0.219 0.151 0.119 0.095 0.092 0.107 0.085 0.081 0.106 0.072 0.047 0.032 19 0.768 0.713 0.644 0.528 0.599 0.807 1.009 1.035 0.750 0.722 0.790 0.907 0.921 0.866 0.924 0.375 0.219 0.144 0.118 0.116 0.123 0.120 0.114 0.119 0.115 0.096 0.070 20 0.811 0.655 0.593 0.496 0.601 0.694 0.810 0.835 0.669 0.671 0.718 0.641 0.566 0.524 0.573 0.290 0.182 0.148 0.123 0.119 0.141 0.103 0.098 0.130 0.080 0.051 0.036 21 0.827 0.714 0.660 0.535 0.601 0.660 0.729 0.775 0.619 0.601 0.640 0.667 0.571 0.525 0.602 0.242 0.160 0.120 0.103 0.122 0.164 0.127 0.133 0.182 0.105 0.059 0.037 22 0.673 0.492 0.427 0.447 0.584 0.751 0.908 0.931 0.656 0.611 0.623 0.552 0.466 0.418 0.443 0.275 0.199 0.146 0.104 0.094 0.106 0.074 0.070 0.095 0.064 0.042 0.030 23 0.949 0.852 0.711 0.559 0.586 0.701 0.855 0.907 0.737 0.731 0.801 0.956 0.934 0.881 0.934 0.380 0.239 0.158 0.127 0.125 0.138 0.126 0.124 0.138 0.118 0.093 0.063 24 0.939 0.835 0.723 0.622 0.716 0.791 0.908 1.031 0.894 0.948 1.036 1.079 0.948 0.897 0.983 0.386 0.260 0.189 0.150 0.160 0.199 0.155 0.163 0.219 0.145 0.101 0.070 25 1.055 0.989 0.894 0.681 0.663 0.726 0.837 0.914 0.813 0.840 0.916 0.892 0.837 0.785 0.846 0.359 0.237 0.179 0.151 0.150 0.175 0.145 0.128 0.147 0.116 0.086 0.058
O separador de campo é espaço e o separador decimal é ponto (.). Além disso foi retirado o parâmetro skiprows = 1, pois a primeira linha, que indica o comprimento de onda, será útil para localizar a coluna de interesse.
data_espectros = pd.read_csv ('tabela_25_espectros_UV_Vis_25_comp_onda_220_350_de_mist_10_PAH.csv', sep = ' ', decimal=b'.', skipinitialspace=True)
print(data_espectros)
E a saída no terminal é:
nm 220 225 230 235 240 245 250 255 260 ... 305 310 315 320 325 330 335 340 345 350
0 1 0.771 0.714 0.658 0.537 0.587 0.671 0.768 0.837 0.673 ... 0.161 0.135 0.137 0.162 0.130 0.127 0.165 0.110 0.075 0.053
1 2 0.951 0.826 0.737 0.638 0.738 0.911 1.121 1.162 0.869 ... 0.185 0.157 0.159 0.180 0.155 0.150 0.178 0.140 0.105 0.077
2 3 0.912 0.847 0.689 0.514 0.504 0.622 0.805 0.892 0.697 ... 0.168 0.134 0.123 0.136 0.115 0.095 0.102 0.089 0.068 0.048
3 4 0.688 0.679 0.662 0.558 0.655 0.738 0.838 0.883 0.670 ... 0.172 0.138 0.144 0.179 0.130 0.134 0.191 0.107 0.060 0.046
4 5 0.873 0.801 0.732 0.640 0.750 0.820 0.907 0.955 0.692 ... 0.170 0.151 0.162 0.201 0.158 0.170 0.239 0.146 0.094 0.067
5 6 0.953 0.850 0.732 0.612 0.724 0.882 1.013 1.087 0.860 ... 0.175 0.138 0.141 0.167 0.129 0.135 0.178 0.115 0.078 0.056
6 7 0.613 0.577 0.547 0.448 0.508 0.463 0.473 0.549 0.510 ... 0.165 0.136 0.142 0.183 0.122 0.129 0.193 0.089 0.041 0.030
7 8 0.927 0.866 0.799 0.605 0.618 0.671 0.771 0.842 0.646 ... 0.155 0.120 0.123 0.144 0.122 0.127 0.164 0.113 0.078 0.056
8 9 0.585 0.577 0.543 0.450 0.515 0.614 0.734 0.815 0.624 ... 0.167 0.136 0.126 0.137 0.109 0.104 0.129 0.098 0.074 0.057
9 10 0.835 0.866 0.803 0.550 0.563 0.577 0.642 0.732 0.675 ... 0.173 0.153 0.160 0.186 0.156 0.157 0.193 0.134 0.093 0.066
10 11 0.477 0.454 0.450 0.433 0.538 0.593 0.661 0.724 0.554 ... 0.139 0.114 0.118 0.154 0.100 0.100 0.154 0.071 0.030 0.016
11 12 0.496 0.450 0.402 0.331 0.389 0.504 0.613 0.599 0.383 ... 0.094 0.076 0.070 0.077 0.065 0.056 0.065 0.053 0.036 0.025
12 13 0.594 0.512 0.441 0.392 0.489 0.562 0.637 0.647 0.415 ... 0.076 0.062 0.076 0.110 0.082 0.094 0.144 0.078 0.043 0.028
13 14 0.512 0.478 0.409 0.319 0.375 0.395 0.436 0.487 0.439 ... 0.122 0.089 0.087 0.107 0.075 0.079 0.114 0.064 0.040 0.028
14 15 0.662 0.583 0.586 0.564 0.687 0.708 0.748 0.757 0.611 ... 0.133 0.113 0.128 0.168 0.125 0.143 0.211 0.114 0.067 0.044
15 16 0.768 0.588 0.501 0.386 0.429 0.539 0.671 0.740 0.607 ... 0.116 0.093 0.089 0.095 0.087 0.081 0.087 0.081 0.069 0.047
16 17 0.635 0.557 0.487 0.377 0.404 0.488 0.597 0.669 0.603 ... 0.139 0.105 0.096 0.104 0.084 0.071 0.077 0.061 0.045 0.031
17 18 0.575 0.489 0.432 0.375 0.408 0.449 0.525 0.569 0.442 ... 0.119 0.095 0.092 0.107 0.085 0.081 0.106 0.072 0.047 0.032
18 19 0.768 0.713 0.644 0.528 0.599 0.807 1.009 1.035 0.750 ... 0.144 0.118 0.116 0.123 0.120 0.114 0.119 0.115 0.096 0.070
19 20 0.811 0.655 0.593 0.496 0.601 0.694 0.810 0.835 0.669 ... 0.148 0.123 0.119 0.141 0.103 0.098 0.130 0.080 0.051 0.036
20 21 0.827 0.714 0.660 0.535 0.601 0.660 0.729 0.775 0.619 ... 0.120 0.103 0.122 0.164 0.127 0.133 0.182 0.105 0.059 0.037
21 22 0.673 0.492 0.427 0.447 0.584 0.751 0.908 0.931 0.656 ... 0.146 0.104 0.094 0.106 0.074 0.070 0.095 0.064 0.042 0.030
22 23 0.949 0.852 0.711 0.559 0.586 0.701 0.855 0.907 0.737 ... 0.158 0.127 0.125 0.138 0.126 0.124 0.138 0.118 0.093 0.063
23 24 0.939 0.835 0.723 0.622 0.716 0.791 0.908 1.031 0.894 ... 0.189 0.150 0.160 0.199 0.155 0.163 0.219 0.145 0.101 0.070
24 25 1.055 0.989 0.894 0.681 0.663 0.726 0.837 0.914 0.813 ... 0.179 0.151 0.150 0.175 0.145 0.128 0.147 0.116 0.086 0.058
[25 rows x 28 columns]
Vamos extrair os valores de absorbância em 335nm e armazenar na variável array_abs_335, do tipo array, com os comandos:
data_abs_335 = data_espectros['335']
array_abs_335 = data_abs_335.values[:]
array_abs_335 = array_abs_335.reshape(-1,1)
print("array_abs_335 : \n", array_abs_335)
E temos a saída:
array_abs_335 : [[0.165] [0.178] [0.102] [0.191] [0.239] [0.178] [0.193] [0.164] [0.129] [0.193] [0.154] [0.065] [0.144] [0.114] [0.211] [0.087] [0.077] [0.106] [0.119] [0.13 ] [0.182] [0.095] [0.138] [0.219] [0.147]]
A seleção da coluna também poderia ser feita com o comando data_abs_335 = data_espectros.loc[:,['335']].
Para exibir o gráfico da figura U.2 com as leituras de absorbância em 335nm das 25 misturas de PAH em função da concentração de pireno (Py), usamos os seguintes comandos:
#Exibindo os dados em gráfico
#conc_min = array_conc_py.min()
conc_max = array_conc_py.max()
#abs_min = array_abs_335.min()
abs_max = array_abs_335.max()
plt.axis([0, conc_max * 1.2, 0, abs_max * 1.2])
plt.xlabel('Concentração de Pireno (mg/L)')
plt.ylabel('Absorbância em 335nm')
#Lista de marcadores
#https://matplotlib.org/3.1.1/api/markers_api.html
plt.plot(array_conc_py, array_abs_335, '.', c='red')
plt.show()
Figura U.2. Gráfico da absorbância em 335nm das 25 misturas de PAH em função da concentração de pireno (Py)
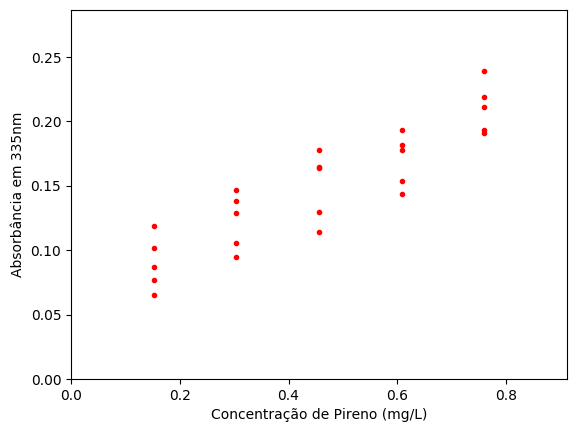
O gráfico mostra uma tendência linear das leituras de aborbância em função da concentração de pireno, e as variações de leitura se devem às contribuições dos demais componentes das misturas.
Vamos usar agora a equação U.1 e calcular a estimativa do parâmetro β1 da equação de regressão, que neste caso é representado por “b”.
Para lembrar:
B ≈ (CtC)-1 CtY
Para simplificar a leitura dos nomes das variáveis vamos substituir os nomes das variáveis array_conc_py e array_abs_335.
conc_py = array_conc_py abs_py = array_abs_335
E portanto a equação é escrita como:
b ≈ ( conc_pyt conc_py )-1 conc_pyt abs_py
A primeira etapa é calcular a transposta do vetor “c”, que é representado pela variável conc_py:
#Transposta do vetor conc_py conc_py_t = conc_py.T
O produto matricial (ctc), obtido pelo produto de conc_py_t por conc_py:
#Transposta do vetor conc_py conc_py_t = conc_py.T #Produto matricial conc_py_t_dot_conc_py = np.dot(conc_py_t, conc_py)
A matriz inversa do produto de conc_py_t por conc_py é:
#Matriz inversa inv_conc_py_t_dot_conc_py = np.linalg.inv(conc_py_t_dot_conc_py) print(inv_conc_py_t_dot_conc_py)
O segundo termo da equação U.1 (cty) pode ser calculado pelo comando:
#Segundo termo da equação
#conc_pyt abs_py
conc_py_t_dot_abs_py = np.dot(conc_py_t, abs_py)
print("conc_py_t_dot_abs_py: ", conc_py_t_dot_abs_py)
E finalmente o escalar “s” que relaciona as leituras de absorbâncias com as concentrações do pireno (Py):
b = np.dot(inv_conc_py_t_dot_conc_py, conc_py_t_dot_abs_py)
print("b: ", b)
b : [[0.30148325]]
Portanto a equação de calibração (modelo empírico) que relaciona a absorbância à concentração de pireno é dado pela equação:
abs_py_est = 0,301 × conc_py
Onde “abs_py_est” corresponde ao valor de absorbância “estimada” pelo modelo de calibração para cada valor de concentração de pireno (conc_py), e descreve uma reta, que parte da origem, como mostra a figura figura S.7
Figura U.3. Gráfico da absorbância em 335nm das 25 misturas de PAH em função da concentração de pireno (Py) com a respectiva reta de regressão pela calibração clássica.
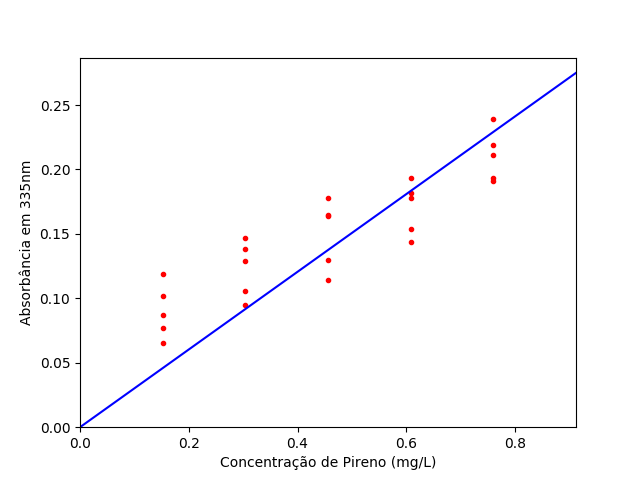
Comandos para a exibição do gráfico com a reta de regressão:
#Exibindo os dados em gráfico incluindo a reta de regressão
#https://scriptverse.academy/tutorials/python-matplotlib-plot-straight-line.html
#conc_min = array_conc_py.min()
conc_max = array_conc_py.max()
#abs_min = array_abs_335.min()
abs_max = array_abs_335.max()
x = np.linspace(0 , 1, 5)
y = b * x
#Convertendo y (matriz coluna 2D) para 1D para ter a mesma dimensionalidade de x
#https://stackoverflow.com/questions/13730468/from-nd-to-1d-arrays
y = y.flatten()
print(x,y)
plt.axis([0, conc_max * 1.2, 0, abs_max * 1.2])
plt.xlabel('Concentração de Pireno (mg/L)')
plt.ylabel('Absorbância em 335nm')
plt.plot(array_conc_py, array_abs_335, '.', c='red')
plt.plot(x, y , '-', c='blue')
plt.show()
Tabela com os valores de concentração, absorbância e absorbância estimada:
Concentração Abs medida Abs estimada
0 0.456 0.165 0.137476
1 0.456 0.178 0.137476
2 0.152 0.102 0.045825
3 0.760 0.191 0.229127
4 0.760 0.239 0.229127
5 0.608 0.178 0.183302
6 0.760 0.193 0.229127
7 0.456 0.164 0.137476
8 0.304 0.129 0.091651
9 0.608 0.193 0.183302
10 0.608 0.154 0.183302
11 0.152 0.065 0.045825
12 0.608 0.144 0.183302
13 0.456 0.114 0.137476
14 0.760 0.211 0.229127
15 0.152 0.087 0.045825
16 0.152 0.077 0.045825
17 0.304 0.106 0.091651
18 0.152 0.119 0.045825
19 0.456 0.130 0.137476
20 0.608 0.182 0.183302
21 0.304 0.095 0.091651
22 0.304 0.138 0.091651
23 0.760 0.219 0.229127
24 0.304 0.147 0.091651
A tabela acima foi montada com o comando:
tabela = np.hstack((conc_py, abs_py, abs_py_est)) #ou com chaves "[]" #tabela = np.hstack([conc_py, abs_py, abs_py_est]) tabela_array = np.hstack([conc_py, abs_py, abs_py_est]) print(tabela_array) tabela_dataframe = pd.DataFrame(data=tabela[:,:], columns=['Concentração', 'Abs medida', 'Abs estimada']) print(tabela_dataframe)
A “qualidade” ou grau de incerteza do modelo de calibração pode ser quantificada pelos “resíduos” (erros) que é a diferença entre os valores observados (experimentais) e os valores calculados (estimados) pelo modelo (abs_py - abs_py_est), expresso geralmente pela “Raiz do Erro Médio Quadrático” (Root Mean Square Error - RMSE), calculado pela equação U.2.
Equação U.2. Equação para o cálculo da Raiz do Erro Médio Quadrático (Root Mean Square Error - RMSE) do modelo.
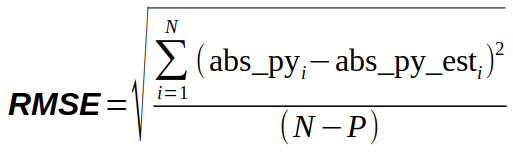
Onde abs_pyi é a absorbância observada para a amostra “i”, abs_py_esti é a absorbância estimada (calculada) pelo modelo para a amostra “i”, N é o número de amostras (25) e P é o número de parâmetros do modelo (1).
Dica
Importante lembrar que o Erro Médio Quadrático é um dos termos necessários para a Tabela de análise de variância (ANOVA).
A Raiz do Erro Médio Quadrático (RMSE em Inglês) pode ser calculada com os comandos:
#Cálculo da Raiz do Erro Médio Quadrático (RMSE) #Valores de absorbância estimada abs_py_est = s * conc_py #Soma Quadrática dos Resíduos SQ_r = np.sum( (abs_py_est - abs_py)**2 ) #Número de amostras (n) n = abs_py.shape[0] #Número de parâmetros (p) p = s.size #Graus de liberdade (gl) gl = n - p #Importando a biblioteca math para o cálculo da raiz import math #Cálculo da Raiz do Erro Médio Quadrático (rmse) rmse = math.sqrt(SQ_r / gl)
Resultando em:
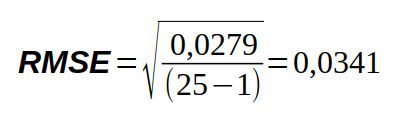
O qual pode ser representado como uma percentagem do valor médio (RMSE% = 100 × (RMSE / abs_py_medio)):
rmse_porcent = ( rmse / np.mean(abs_py))* 100
Que resulta em 22,9%.
Nota
Às vezes, o erro percentual é calculado em relação ao desvio padrão e não à média: isso é mais apropriado se os dados estiverem centrados na média (porque a média é 0).
Mas para estimar a concentração de pireno em uma amostra desconhecida, a equação de calibração deveria ser usada de uma forma “invertida”:
ĉ = 1/b × y
concentração = 3,32 × absorbância
Então podemos fazer a seguinte pergunta: Por que não fazer uma “calibração inversa”?
Essa é o assunto da próxima seção.
Nota
Os comandos usados nesta seção estão organizados em um único arquivo disponível no link: calibracao_univariada.py.
Embora a “calibração clássica univariada” é comumente feita na maioria dos laboratórios, ela não é necessariamente a forma mais apropriada.
Se o objetivo final é estimar a concentração (c) a partir das medidas de absorbância (y), ou outro sinal analítico, podemos fazer a calibração, desde o início, correlacionando a medida espectroscópica (ou outro sinal analítico) à concentração conforme a relação:
Ĉ = Y × B
Outra questão é:
Qual a principal “fonte de incerteza” em uma calibração?
Os erros do sinal analítico (absorbância, área de sinal cromatográfico) são dependentes do instrumento analítico usado. Os quais, ao longo dos anos, têm se tornado cada vez mais “reprodutíveis”.
Mas a concentração (variável independente) é determinada por procedimentos que envolvem: pesagem, medidas volumétricas, diluições e geralmente são “ainda” fontes significativas de incertezas (erros). Pois a qualidade da vidraria volumétrica (balões, pipetas, seringas) não tem avançado muito ao longo dos anos, ao contrário da sensibilidade e reprodutibilidade dos instrumentos analíticos. (Fonte: Chemometrics - Data Driven Extraction for Science, 2018)
A “calibração clássica” (ou convencional) ajusta um modelo no qual se assume que toda a incerteza está na resposta analítica, mas seria mais adequado considerar que os erros estão basicamente nas medidas de concentração, como mostra a figura U.4.
Figura U.4. Principais fontes de erro (incerteza) na calibração clássica (a) e inversa (b) (Fonte: Chemometrics - Data Driven Extraction for Science, 2018)
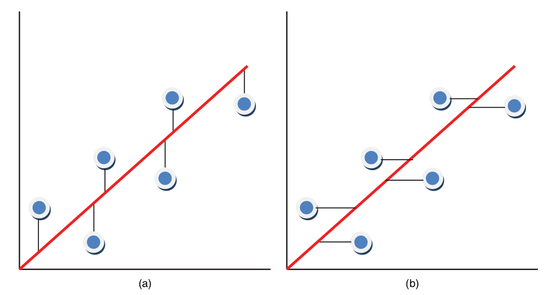
Portanto a calibração inversa é feita conforme a relação:
C ≈ Y × B
Onde, $times; representa o “produto matricial” e “Y” é um vetor coluna formado pelas respostas do sensor (Ex: absorbância em um comprimento de onda) para um conjunto de amostras que formam o vetor “C” que contêm as concentrações do analito de interesse em diferentes concentrações. E “B” é um “escalar” que correlaciona os dois vetores (Y e C) e é determinado pela técnica de Regressão.
Ambos os vetores (Y e C) possuem o mesmo número de linhas correspondente ao número de amostras.
O escalar “b” é determinado pela equação U.4.
Equação U.4. Equação matricial geral para o ajuste de modelos para calibração pela técnica dos “mínimos quadrados”.
B ≈ (YtY)-1 YtC
Aplicando esse modelo aos 25 espectros disponíveis no link tabela_25_espectros_UV_Vis_25_comp_onda_220_350_de_mist_10_PAH.csv referentes às 25 soluções contendo uma mistura de 10 PAHs (disponível para download no link tabela_25_solucoes_10_PAH.csv), obtemos a seguinte equação de calibração que relaciona a concentração à absorbância de pireno:
conc_py_est = 3,16 × abs_py
Figura U.5. Gráfico da absorbância em 335nm das 25 misturas de PAH em função da concentração de pireno (Py) com as respectivas retas de regressão pela calibração clássica (azul) e inversa (verde).
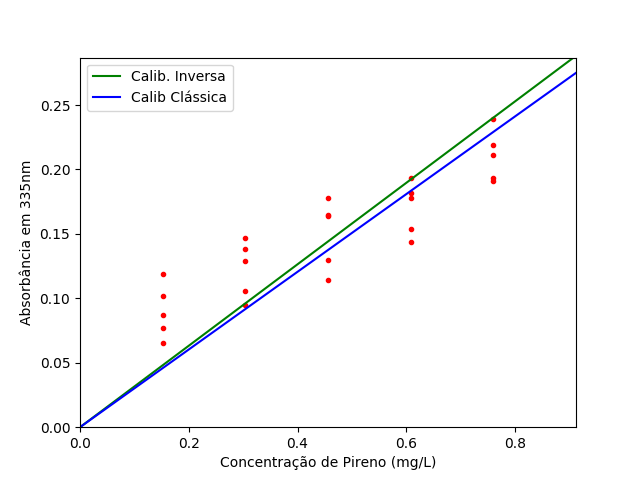
Os cálculos foram realizados com os comandos “adicionais”:
############################################################################
#Regressão inversa
#Transposta do vetor abs_py
abs_py_t = abs_py.T
print(abs_py_t)
#Produto matricial
abs_py_t_dot_abs_py = np.dot(abs_py_t, abs_py)
print(abs_py_t_dot_abs_py)
#Matriz inversa
inv_abs_py_t_dot_abs_py = np.linalg.inv(abs_py_t_dot_abs_py)
print(inv_abs_py_t_dot_abs_py)
#Verificação
identidade = np.dot(abs_py_t_dot_abs_py, inv_abs_py_t_dot_abs_py )
print(identidade)
#Segundo termo da equação
#abs_pyt conc_py
abs_py_t_dot_conc_py = np.dot(abs_py_t, conc_py)
print("abs_py_t_dot_conc_py: " , abs_py_t_dot_conc_py, abs_py_t_dot_conc_py.shape)
b_i = np.dot(inv_abs_py_t_dot_abs_py, abs_py_t_dot_conc_py)
print("b_i: ", b_i)
############################################################################
#Exibindo os dados em gráfico incluindo a reta de regressão
#https://scriptverse.academy/tutorials/python-matplotlib-plot-straight-line.html
#conc_min = array_conc_py.min()
conc_max = conc_py.max()
#abs_min = array_abs_335.min()
abs_max = abs_py.max()
y_i = np.linspace(0 , 0.5, 5)
x_i = b_i * y_i
#Convertendo y (matriz coluna 2D) para 1D para ter a mesma dimensionalidade de x
#https://stackoverflow.com/questions/13730468/from-nd-to-1d-arrays
x_i = x_i.flatten()
print("x_i,y_i :\n", x_i,y_i)
plt.axis([0, conc_max * 1.2, 0, abs_max * 1.2])
plt.xlabel('Concentração de Pireno (mg/L)')
plt.ylabel('Absorbância em 335nm')
plt.plot(conc_py, abs_py, '.', c='red')
plt.plot(x_i, y_i , '-', c='green', label='Calib. Inversa')
plt.plot(x, y, '-', c='blue', label='Calib Clássica')
plt.legend()
plt.show()
Em muitas situações é conveniente incluir um termo extra no modelo de calibração para indicar o ponto de interseção da reta de calibração com o eixo y, gerando um modelo de calibração inversa como na equação U.5
A qual pode ser expresso em notação matricial como:
Onde “C” é um vetor coluna com as concentrações, “B” é um vetor coluna com dois números, o primeiro referente a b0 (interseção) e o segundo referente a b1 (inclinação). E “Y” é a matriz das “ABSORBÂNCIAS”, agora com “2” colunas, sendo a primeira uma coluna de “uns” e a segunda coluna com as absorbâncias.
E neste caso vamos usar “novamente” a Equação matricial geral para o ajuste de modelos de calibração inversa, ou seja:
B ≈ (YtY)-1 YtC
Para calcular os coeficientes do modelo e exibir a reta correspondete adicionamos os seguintes comandos em Python ao código original:
###################################################################################################
#Cálculo da matriz de coeficientes (interseção e inclinação) do modelo de calibração inversa
abs_py = np.hstack([np.ones(abs_py.shape), abs_py])
print("abs_py e formato: \n", abs_py, abs_py.shape)
#Transposta do vetor abs_py
abs_py_t = abs_py.T
print()
print("abs_py_t e formato: \n", abs_py_t, abs_py_t.shape)
print()
#Produto matricial
abs_py_t_dot_abs_py = np.dot(abs_py_t, abs_py)
print()
print("abs_py_t_dot_abs_py: \n", abs_py_t_dot_abs_py)
#Matriz inversa
inv_abs_py_t_dot_abs_py = np.linalg.inv(abs_py_t_dot_abs_py)
print("inv_abs_py_t_dot_abs_py: \n", inv_abs_py_t_dot_abs_py)
#Verificação
identidade = np.dot(abs_py_t_dot_abs_py, inv_abs_py_t_dot_abs_py )
print("identidade :", identidade)
#Segundo termo da equação
#abs_pyt conc_py
print("abs_py_t e formato: \n", abs_py_t, abs_py_t.shape)
print()
abs_py_t_dot_conc_py = np.dot(abs_py_t, conc_py)
print("abs_py_t_dot_conc_py: " , abs_py_t_dot_conc_py, abs_py_t_dot_conc_py.shape)
b_i = np.dot(inv_abs_py_t_dot_abs_py, abs_py_t_dot_conc_py)
print("b_i: ", b_i)
#Exibindo a reta com interseção
#y_i_int = lista com os valores de absorbância no intervalo de 0 - 0.5
y_i_int = np.linspace(0 , 0.5, 5)
#Mas os valores de absorbância precisam estar no formato matricial com 2 colunas
#sendo a primeira coluna de uns
y_i_int = y_i_int.reshape(-1,1)
y_i_int_uns = np.hstack([np.ones(y_i_int.shape), y_i_int])
print("y_i_int_uns e shape: \n", y_i_int_uns, y_i_int_uns.shape)
print("b_i e shape: \n", b_i, b_i.shape)
x_i_int = np.dot(y_i_int_uns, b_i)
print("x_i_int e shape: \n", x_i_int, x_i_int.shape)
#Convertendo y (matriz coluna 2D) para 1D para ter a mesma dimensionalidade de x
#https://stackoverflow.com/questions/13730468/from-nd-to-1d-arrays
x_i_int = x_i_int.flatten()
y_i_int = y_i_int.flatten()
print("x_i_int , y_i_int :\n", x_i_int, y_i_int)
plt.axis([0, conc_max * 1.2, 0, abs_max * 1.2])
plt.xlabel('Concentração de Pireno (mg/L)')
plt.ylabel('Absorbância em 335nm')
plt.plot(conc_py, abs_py, '.', c='red')
plt.plot(x_i_int, y_i_int, '-', c='black', label='Calib. Inversa com Interseção')
plt.plot(x_i, y_i , '-', c='green', label='Calib. Inversa')
plt.plot(x, y, '-', c='blue', label='Calib Clássica')
plt.legend()
plt.show()
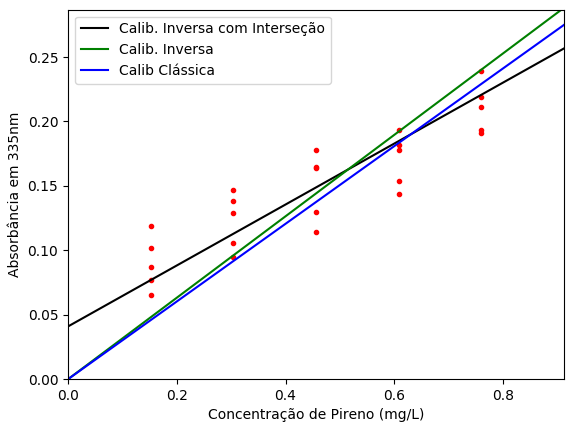
E obtemos a seguinte equação de calibração que relaciona a concentração à absorbância de pireno:
conc_py_est = -0,17 + 4,23 × abs_py
Gerando o gráfico da figura U.6.
Figura U.6. Gráfico da absorbância em 335nm das 25 misturas de PAH em função da concentração de pireno (Py) com as respectivas retas de regressão pela calibração clássica (azul), inversa (verde) e com interseção (preto).
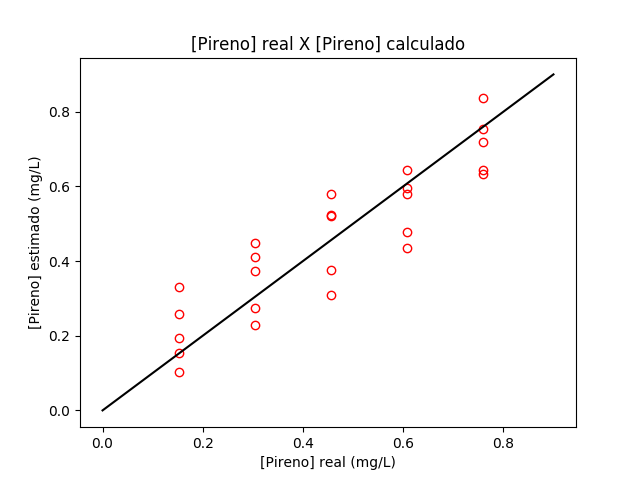
E o gráfico da figura U.7 mostra as concentrações estimadas pela equação de calibração em comparação com as concentrações reais de pireno.
Figura U.7. Gráfico das concentrações estimadas de pireno em relação às concentrações reais, obtidas pela técnica de Calibração Univariada Inversa com Interseção, usando 1 comprimento de onda (330nm) para a calibração.
Nota
Os comandos usados nesta seção estão organizados em um único arquivo disponível no link: calibracao_univariada_inversa.py.
Na calibração univariada não há de fato uma diferença significativa entre a calibração clássica e a calibração inversa.
Mas na “calibração multivariada” essa diferença é “muito importante”, como será visto mais adiante.