Nessa seção, vamos discutir o uso da Estatística Multivariada na “Calibração Multivariada” em Química Analítica.
Nota
As principais fontes de informação sobre Calibração Multivariada em Química Analítica foram: Introduction to multivariate calibration in analytical chemistry, 2000 e Introduction to Multivariate Calibration - A Practical Approach, 2018.
Na “calibração multivariada” são feitas várias leituras de dados para cada amostra. Por exemplo, diferentes valores de absorção, na região UV/Vis, ou emissão de fluorescência, para cada amostra contendo uma substância com concentração conhecida.
Mas quais as vantagens da Calibração Multivariada em relação à Calibração Univariada?
A resposta é: “interferentes”.
Na química analítica clássica, qualquer substância que produza um sinal semelhante ao sinal do analito de interesse (por exemplo, absorvendo no mesmo comprimento de onda) é considerada um “interferente”.
Algumas das possíveis alternativas para evitar o efeito indesejado dos interferentes:
remover fisicamente o constituinte interferente por um procedimento de purificação (Ex: filtração, precipitação),
separar os constituintes por cromatografia ou outras técnicas separativas (Ex: destilação),
mascarar o interferente pela reação com uma reagente específico que transforme o interferente em um produto inerte,
usar um reagente específico para transformar o analito em um produto que mostre um sinal diferente dos interferentes etc
Mas com a “calibração multivariada” o efeito negativo dos interferentes pode ser compensado sem a necessidade de removê-los ou transformá-los, como na calibração univariada.
Dúvidas mais frequentes sobre calibração multivariada:
É necessário conhecer todos os possíveis interferentes que podem estar presentes em uma amostra desconhecida para aplicar um protocolo de calibração multivariada?
Em geral, não; na maioria das aplicações, a identidade química dos interferentes não é conhecida.
Como preparar um conjunto de amostras de calibração contendo os interferentes, se eles não são conhecidos?
O conjunto de calibração deve conter um número de amostras representativas, contendo quantidades variadas dos interferentes, mesmo quando os interferentes não forem conhecidos. Somente o conteúdo do analito de interesse precisa ser conhecido nas amostras de calibração, seja porque uma quantidade conhecida do analito “puro” (padrão) foi usada para preparar as amostras ou porque o conteúdo do analito foi medido por uma técnica analítica de referência.
É necessário preparar um grande número de amostras de calibração?
Esta é uma pergunta muito importante, mas sem uma resposta simples. Em geral, recomenda-se um número de amostras na ordem de “centenas”. O conteúdo do analito deve ser conhecido ou medido em todas essas amostras. O modelo de calibração deverá ser “validado” contra outro conjunto independente de amostras com conteúdo conhecido do analito. Essa fase de validação permite avaliar se o modelo é satisfatório ou não, dependendo do erro médio de previsão para o conjunto de amostras de validação. Pode ser necessário aumentar o número de amostras de calibração (treino) se o erro médio de previsão for grande, até que o modelo multivariado se estabilize, fornecendo um erro de previsão considerado como “aceitável”.
Mas o que é considerado como um erro de previsão “aceitável”?
A grosso modo, o erro médio de previsão (em % em relação à concentração média do analito usado na calibração) pode ser caracterizado como “excelente” se for menor que 2%, “bom” se estiver na faixa de 2 a 5%, “razoável” se estiver na faixa de 5 a 10% e “ruim” se maior que 10%. No entanto, há um fator importante a ser levado em consideração: um método recém-desenvolvido deve competir com os métodos existentes, não apenas em termos de erro relativo de previsão, mas também em termos de custo, velocidade e simplicidade. É um compromisso entre esses parâmetros, e possivelmente outros, o que determina se uma calibração multivariada é capaz de competir favoravelmente ou não. Por exemplo, se o único método disponível tem um erro associado de 15% e o modelo de calibração desenvolvido mostra um erro relativo de 10%, então o modelo multivariado já não pode ser considerado “ruim”!
Qual o “prazo de validade” de um modelo de calibração multivariado?
Em alguns instrumentos, particularmente nos espectrômetros NIR, pode haver alterações na resposta do detector ou nas condições de medição com o tempo. Por outro lado, não há garantias de que as amostras recém-produzidas ou coletadas sempre tenham a mesma composição química, especialmente quando as amostras são de origem natural. De qualquer forma, os modelos multivariados têm a capacidade de sinalizar se essas amostras possuem composição diferente em relação ao conjunto de calibração. Essa propriedade importante será explorada no futuro e é conhecida como vantagem de “primeira ordem”.
Vantagem de primeira ordem significa a possibilidade de realizar análises mesmo na presença de interferentes, desde que esses interferentes estejam presentes nas amostras de calibração.
O que fazer se novas amostras estiverem fora da faixa de calibração ou contiverem constituintes químicos que não foram considerados na fase de calibração?
Esta pergunta se refere a duas questões independentes. Por um lado, se as composições qualitativas das amostras de calibração e teste forem análogas, mas se o analito estiver presente nas amostras de teste em concentrações fora da faixa de calibração, o modelo ainda poderá ser útil. Isso será possível se a relação entre o sinal e a concentração do analito for linear e se a linearidade se estender além da faixa de calibração.
Por outro lado, se as amostras de teste contiverem novos constituintes químicos, deve-se recalibrar o modelo, incluindo várias novas amostras no conjunto de calibração, com a concentração conhecida do analito, para recuperar a representatividade e permitir que o modelo se adapte às novas condições. Os laboratórios industriais geralmente verificam a calibração com uma certa frequência (uma vez por mês, semestre, ano etc.) usando um conjunto de amostras com concentração conhecida de analito, o que significa que os métodos analíticos de referência não devem ser descartados e devem ser mantidos para esse controle periódico da capacidade do modelo.
Por exemplo, um laboratório que controla a qualidade do caldo de cana-de-açúcar desenvolveu um modelo multivariado para medir o grau Brix (uma medida do conteúdo de carboidratos na cana) usando espectroscopia NIR. Um grande número de amostras de cana-de-açúcar foi empregado para calibração, medindo os espectros NIR e o grau Brix com um polarímetro (o método de referência), obtendo um modelo razoavelmente estável com bons parâmetros analíticos. No entanto, em um ano com frio extremo na região produtora de cana, o modelo perdeu “representatividade”. O conjunto de amostras de controle começou a mostrar resultados analíticos ruins e isso levou à recalibração do modelo. A solução foi adicionar, ao conjunto de calibração original, centenas de novos caldos de cana de açúcar da estação fria. O modelo se estabilizou novamente com um erro de previsão razoável. As condições futuras de clima frio não deveriam afetar o modelo de calibração.
Existem muitas armadilhas no uso de modelos de calibração, talvez a mais grave seja a variabilidade no desempenho de um instrumento ao longo do tempo. Cada instrumento tem características diferentes e a resposta pode variar durante o dia ou até em algumas horas. Às vezes, é necessário refazer o modelo de calibração regularmente, executando um conjunto de amostras de calibração, possivelmente diariamente ou semanalmente.
Aplicações da Calibração Multivariada:
O caso mais simples é a calibração da concentração de um único composto usando um método espectroscópico ou cromatográfico. Por exemplo, a determinação da concentração de clorofila por espectroscopia de absorção. Nesse caso, ao invés de usar apenas um comprimento de onda (como na calibração univariada), a calibração multivariada envolve o uso de vários comprimentos de onda.
Um caso mais complexo é o de uma mistura de vários componentes, onde todos os componentes puros estão presentes no conjunto de amostras de calibração, por exemplo, em uma mistura de quatro produtos farmacêuticos.
É possível controlar a concentração dos compostos de referência, para que várias misturas cuidadosamente projetadas possam ser produzidas em laboratório. Às vezes, o objetivo é verificar se o espectro de uma mistura pode ser usado para determinar concentrações individuais e, se for o caso, com que grau de confiabilidade.
O objetivo pode ser também substituir um lento e caro método de separação cromatográfica por uma abordagem espectroscópica rápida.
Outro objetivo pode ser o monitoramento de impurezas, e até que ponto a concentração de uma pequena impureza pode ser determinada, por exemplo, encoberta em um grande pico cromatográfico.
Uma abordagem diferente pode ser necessária se, em uma mistura, for conhecida a concentração de apenas uma porção dos componentes da mistura, por exemplo, os hidrocarbonetos poliaromáticos na fração de voláteis do alcatrão de carvão.
Por exemplo, nas amostras naturais, pode haver dezenas ou centenas de componentes desconhecidos, mas apenas algumas podem ser quantificados e calibrados.
Os interferentes desconhecidos não podem ser necessariamente determinados e não é possível projetar um conjunto de amostras em laboratório contendo todos os componentes potenciais em amostras reais.
A calibração multivariada é eficaz, desde que o intervalo de composição das amostras usadas para desenvolver o modelo (calibração) seja suficientemente representativo de todas as futuras amostras a serem analisadas. Caso contrário, as previsões da calibração multivariada poderão ser muito imprecisas.
Para evitar problemas com amostras que não pertencem ao conjunto de dados originais, foram desenvolvidas várias abordagens para a determinação de “outliers” (valores discrepantes, atípicos, anormais).
Um caso final é quando o objetivo da calibração não é determinar a concentração de um composto em particular, mas “um grupo de compostos”, por exemplo, proteína no trigo.
Nesse caso os métodos só funcionarão se estiver disponível um conjunto de amostras suficientemente grande e representativo.
Por exemplo, na química de alimentos, se o fornecedor de um produto for uma fonte conhecida que provavelmente não será alterada, geralmente é adequado configurar um modelo de calibração nesse conjunto de treinamento.
Vamos usar o dataset com 25 espectros de 25 soluções diferentes contendo misturas de 10 hidrocarbonetos poliaromáticos (PAH) com 5 concentrações diferentes.
Os espectros estão disponíveis para download no link tabela_25_espectros_UV_Vis_25_comp_onda_220_350_de_mist_10_PAH.csv.
E a tabela com as 25 amostras com 5 diferentes concentrações dos 10 Hidrocarbonetos Poliaromáticos está disponível para download no link tabela_25_solucoes_10_PAH.csv.
Somente alguns PAH absorvem acima de 330 nm (Pireno, Fluoranteno, Acenaftaleno e Benzo[a]antraceno), e por isso, é possível selecionar “4” comprimentos de onda (330, 335, 340 e 345 nm), nos quais as absorbâncias desses 4 compostos são diferentes.
Py - pyrene (Pireno);
Ace - acenaphthene (Acenaftaleno);
Benz - benzanthracene (Benzo[a]antraceno);
Fluora - fluoranthene (Fluoranteno);
Nota
Para a determinação de N componentes da mistura, é necessário usar “no mínimo” o mesmo número de comprimentos de onda (ou sinais de outra natureza).
Mas isso não impede que sejam usados mais comprimentos de onda para melhorar a qualidade da calibração.
A figura V.1 mostra os espectros de absorção dos 4 compostos puros nos comprimentos de onda que serão usados.
Figura V.1. Em destaque o segmento do espectro com as absorbâncias de 4 PAHs (Pireno, Fluoranteno, Acenaftaleno e Benzo[a]antraceno) em 330, 335, 340 e 345 nm.
![Em destaque o segmento do espectro com as absorbâncias de 4 PAHs (Pireno, Fluoranteno, Acenaftaleno e Benzo[a]antraceno) em 330, 335, 340 e 345 nm.](figuras/abs_Py_Fluor_Benz_Ace_330_335_340_345.png)
Não é necessário que os comprimentos de onda selecionados sejam sequenciais, mas que sejam mais representativos dos 4 compostos analisados e que as intensidades de absorção relativas sejam differentes.
Mas se houver “correlação” entre as concentrações ou entre o perfil dos espectros, pode ser difícil quantificar satisfatóriamente todos os compostos individualmente.
Nota
A falta de uma padronização nos símbolos usados para representar as variáveis “dependentes” e “independentes” costuma gerar confusão na leitura dos textos de Quimiometria.
Nesta seção estou considerando a “Concentração” como a variável “Independente”. E a “Absorbância” o sinal analítico proporcional à Concentração e portanto como variável “Dependente”.
A matriz com as concentrações das amostras é representada pela letra “C” e a matriz do sinal analítico (Absorbância) é representada pela letra “Y”.
Neste caso o sinal analítico é a absorbância, mas poderia representar um sinal analítico de qualquer natureza (fluorescência, potencial elétrico, área de pico cromatográfico) proporcional à concentração do analito.
Em seguida vamos tomar as seguintes providências para a “calibração multivariada inversa”:
selecionar as absorbâncias nos 4 comprimentos de onda escolhidos em todos os 25 espectros e montar a matriz Y,
selecionar as concentrações dos 4 compostos escolhidos em todas as 25 soluções e montar a matriz C,
calcular a matriz de coeficientes B que relaciona Y através da relação C ≈ Y × B (calibração multivariada inversa - equação V.1), onde B é uma matriz 4×4 na qual cada coluna representa um composto e cada linha corresponde a um comprimento de onda. E a matriz B é obtida pela equação V.2
Equação V.2. Equação matricial geral para o cálculo da matriz de parâmetros B pela técnica dos “mínimos quadrados” para a “calibração multivariada inversa”.
B ≈ (Yt • Y)-1 • Yt • C
Comandos Python para a montagem das matrizes C (concentração dos 4 PAHs) e Y (absorbâncias em 330, 335, 340 e 345nm):
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
#Para usar toda a largura do terminal
pd.set_option('display.width', pd.util.terminal.get_terminal_size()[0])
espectros = pd.read_csv ('tabela_25_espectros_UV_Vis_25_comp_onda_220_350_de_mist_10_PAH.csv', sep = ' ', decimal=b'.', skipinitialspace=True)
#print(espectros)
espectros_330_335_340_345 = espectros[['330', '335', '340', '345']]
print("espectros_330_335_340_345 :\n", espectros_330_335_340_345)
Y = espectros_330_335_340_345.values[:]
print("Y :\n", Y)
solucoes = pd.read_csv ('tabela_25_solucoes_10_PAH.csv', sep = ',', skiprows = 1, decimal=b',', skipinitialspace=True)
#print(solucoes)
#Seleciona a coluna referente ao pireno (Py)
solucoes_4_PAH = solucoes[['Py', 'Ace', 'Benz', 'Fluora']]
print(solucoes_4_PAH)
C = solucoes_4_PAH.values[:]
print(C)
Saída do dataframe e da matriz C no terminal:
Py Ace Benz Fluora
0 0.456 0.12 1.62 0.12
1 0.456 0.04 2.70 0.12
2 0.152 0.20 1.62 0.08
3 0.760 0.20 1.08 0.16
4 0.760 0.16 2.16 0.16
5 0.608 0.20 2.16 0.04
6 0.760 0.12 0.54 0.16
7 0.456 0.08 2.16 0.12
8 0.304 0.16 1.62 0.20
9 0.608 0.16 2.70 0.04
10 0.608 0.04 0.54 0.04
11 0.152 0.16 0.54 0.08
12 0.608 0.12 1.08 0.04
13 0.456 0.20 0.54 0.12
14 0.760 0.04 1.62 0.16
15 0.152 0.04 2.16 0.08
16 0.152 0.08 1.08 0.08
17 0.304 0.04 1.08 0.20
18 0.152 0.12 2.70 0.08
19 0.456 0.16 1.08 0.12
20 0.608 0.08 1.62 0.04
21 0.304 0.08 0.54 0.20
22 0.304 0.20 2.70 0.20
23 0.760 0.08 2.70 0.16
24 0.304 0.12 2.16 0.20
C :
[[0.456 0.12 1.62 0.12 ]
[0.456 0.04 2.7 0.12 ]
[0.152 0.2 1.62 0.08 ]
[0.76 0.2 1.08 0.16 ]
[0.76 0.16 2.16 0.16 ]
[0.608 0.2 2.16 0.04 ]
[0.76 0.12 0.54 0.16 ]
[0.456 0.08 2.16 0.12 ]
[0.304 0.16 1.62 0.2 ]
[0.608 0.16 2.7 0.04 ]
[0.608 0.04 0.54 0.04 ]
[0.152 0.16 0.54 0.08 ]
[0.608 0.12 1.08 0.04 ]
[0.456 0.2 0.54 0.12 ]
[0.76 0.04 1.62 0.16 ]
[0.152 0.04 2.16 0.08 ]
[0.152 0.08 1.08 0.08 ]
[0.304 0.04 1.08 0.2 ]
[0.152 0.12 2.7 0.08 ]
[0.456 0.16 1.08 0.12 ]
[0.608 0.08 1.62 0.04 ]
[0.304 0.08 0.54 0.2 ]
[0.304 0.2 2.7 0.2 ]
[0.76 0.08 2.7 0.16 ]
[0.304 0.12 2.16 0.2 ]] (25, 4)
Saída do dataframe e da matriz Y no terminal:
espectros_330_335_340_345 :
330 335 340 345
0 0.127 0.165 0.110 0.075
1 0.150 0.178 0.140 0.105
2 0.095 0.102 0.089 0.068
3 0.134 0.191 0.107 0.060
4 0.170 0.239 0.146 0.094
5 0.135 0.178 0.115 0.078
6 0.129 0.193 0.089 0.041
7 0.127 0.164 0.113 0.078
8 0.104 0.129 0.098 0.074
9 0.157 0.193 0.134 0.093
10 0.100 0.154 0.071 0.030
11 0.056 0.065 0.053 0.036
12 0.094 0.144 0.078 0.043
13 0.079 0.114 0.064 0.040
14 0.143 0.211 0.114 0.067
15 0.081 0.087 0.081 0.069
16 0.071 0.077 0.061 0.045
17 0.081 0.106 0.072 0.047
18 0.114 0.119 0.115 0.096
19 0.098 0.130 0.080 0.051
20 0.133 0.182 0.105 0.059
21 0.070 0.095 0.064 0.042
22 0.124 0.138 0.118 0.093
23 0.163 0.219 0.145 0.101
24 0.128 0.147 0.116 0.086
Y :
[[0.127 0.165 0.11 0.075]
[0.15 0.178 0.14 0.105]
[0.095 0.102 0.089 0.068]
[0.134 0.191 0.107 0.06 ]
[0.17 0.239 0.146 0.094]
[0.135 0.178 0.115 0.078]
[0.129 0.193 0.089 0.041]
[0.127 0.164 0.113 0.078]
[0.104 0.129 0.098 0.074]
[0.157 0.193 0.134 0.093]
[0.1 0.154 0.071 0.03 ]
[0.056 0.065 0.053 0.036]
[0.094 0.144 0.078 0.043]
[0.079 0.114 0.064 0.04 ]
[0.143 0.211 0.114 0.067]
[0.081 0.087 0.081 0.069]
[0.071 0.077 0.061 0.045]
[0.081 0.106 0.072 0.047]
[0.114 0.119 0.115 0.096]
[0.098 0.13 0.08 0.051]
[0.133 0.182 0.105 0.059]
[0.07 0.095 0.064 0.042]
[0.124 0.138 0.118 0.093]
[0.163 0.219 0.145 0.101]
[0.128 0.147 0.116 0.086]] (25, 4)
Cálculo da matriz de coeficientes B segundo a equação matricial V.2:
#Cálculo da matriz de coeficientes B
#Transposta da matriz Y Y_t
Y_t = Y.T
print("Y_t :\n", Y_t)
#Produto matricial Y_t dot Y
Y_t_dot_Y = np.dot(Y_t, Y)
print("Y_t_dot_Y :", Y_t_dot_Y)
#Inversa do produto matricial
inv_Y_t_dot_Y = np.linalg.inv(Y_t_dot_Y)
print("inv_Y_t_dot_Y :\n", inv_Y_t_dot_Y)
#Segundo termo da equação Y^tC
Y_t_dot_C = np.dot(Y_t, C)
print("Y_t_dot_C :\n", Y_t_dot_C)
#Cálculo da matriz de coeficientes B
B = np.dot(inv_Y_t_dot_Y, Y_t_dot_C)
print("B :\n", B)
B é uma matriz 4×4 na qual cada coluna representa um dos PAH correspondente às colunas da matriz C, e cada linha corresponde a um comprimento de onda correspondente às colunas da matriz Y, como mostra a figura V.2.
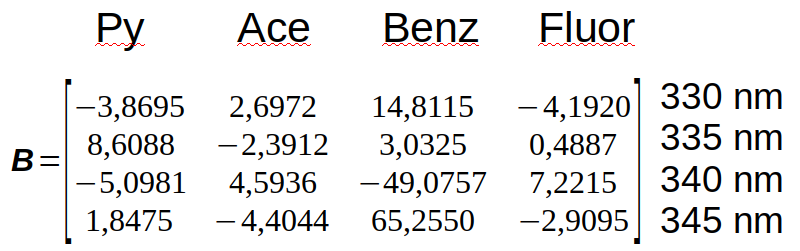
E finalmente as concentrações estimadas de todos os 4 componentes podem ser obtidas pela equação:
Ĉ = YB
O diagrama da figura V.3 mostra a sequência para o cálculo da matriz de concentrações estimadas (Ĉ).
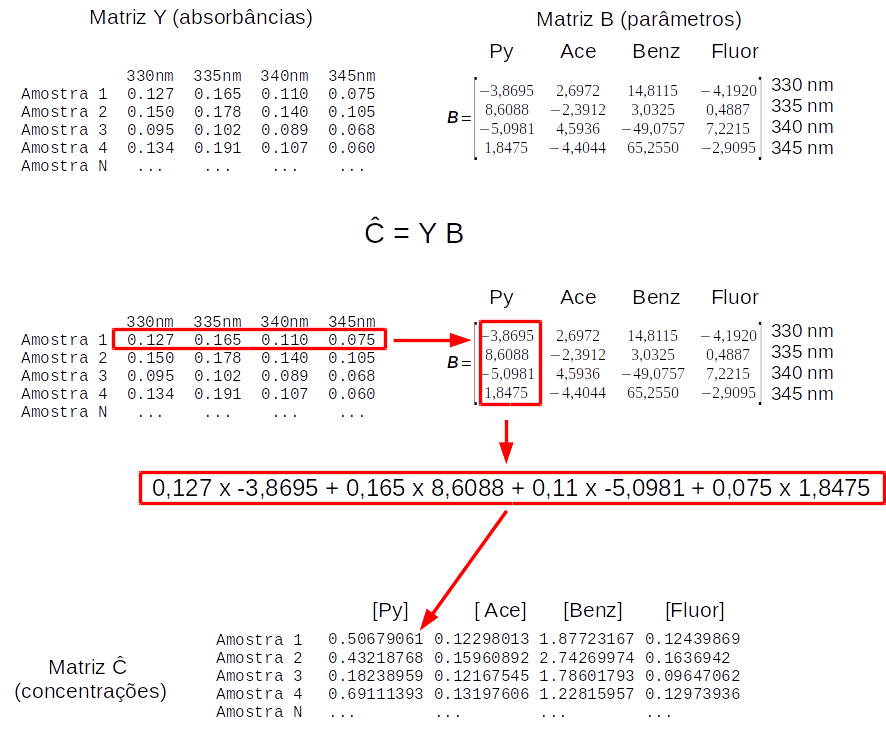
Portanto as equações para o cálculo das concentrações dos 4 PAHs são respectivamente:
[Pireno] = - 3.870 × A330 + 8.609 × A335 - 5.098 × A340 + 1.848 × A345
[Acenaftaleno] = 2.6972 × A330 - 2.3912 × A335 + 4.5936 × A340 - 4.4044 × A345
[Benzantraceno] = 14.8115 × A330 + 3.0325 × A335 - 49.0757 × A340 + 65.2550 × A345
[Fluoranteno] = - 4.1920 × A330 + 0.4887 × A335 + 7.2215 × A340 - 2.9095 × A345
Os coeficientes foram calculados para os comprimentos de onda escolhidos e portanto se forem selecionados outros comprimentos de onda serão obtidos outros valores para a matriz de coeficientes B.
Além disso, as diferenças entre os espectrofotômetros podem fazer com que essa equação não seja válida para todos os instrumentos, e portanto tenha que ser refeita para um instrumento específico.
Comandos Python para estimar as concentrações dos 4 PAHs:
#Cálculo das concentrações dos 4 PAHs
C_est = np.dot(Y, B)
print("C_est :\n", C_est)
Gerando a saída correspondente às concentrações dos 4 PAHs (Pireno, Fluoranteno, Acenaftaleno e Benzo[a]antraceno) nas 25 soluções de calibração:
C_est : [[0.50679061 0.12298013 1.87723167 0.12439869] [0.43218768 0.15960892 2.74269974 0.1636942 ] [0.18238959 0.12167545 1.78601793 0.09647062] [0.69111393 0.13197606 1.22815957 0.12973936] [0.82902199 0.14370359 2.21165496 0.18499824] [0.56780058 0.12322807 1.98553337 0.12459452] [0.78434222 0.11470574 0.80368383 0.07697124] [0.48843008 0.12593898 1.92273717 0.13684584] [0.34520347 0.09630561 1.95104901 0.11947337] [0.54265103 0.16791348 2.40326199 0.13326621] [0.63225942 0.09550346 0.42143821 0.08149831] [0.13918789 0.08052534 0.77472892 0.0750086 ] [0.55771997 0.07812954 0.80702951 0.11448968] [0.42333023 0.05830772 0.98517546 0.07033669] [0.80570991 0.10975268 1.53536935 0.13196878] [0.15006389 0.07862755 1.99102944 0.08714667] [0.16029291 0.08939975 1.22798183 0.04957902] [0.31886849 0.08874985 1.05471788 0.09544833] [0.17439755 0.12838471 2.67016284 0.13142194] [0.4263024 0.0963469 1.24770901 0.08204674] [0.62585292 0.14601644 1.21895157 0.11799974] [0.29828411 0.07065565 0.92476375 0.09296045] [0.27843224 0.13691944 2.53290389 0.12918022] [0.70196368 0.13722145 2.5531847 0.17698011] [0.33769671 0.14783168 2.26080881 0.12273429]]
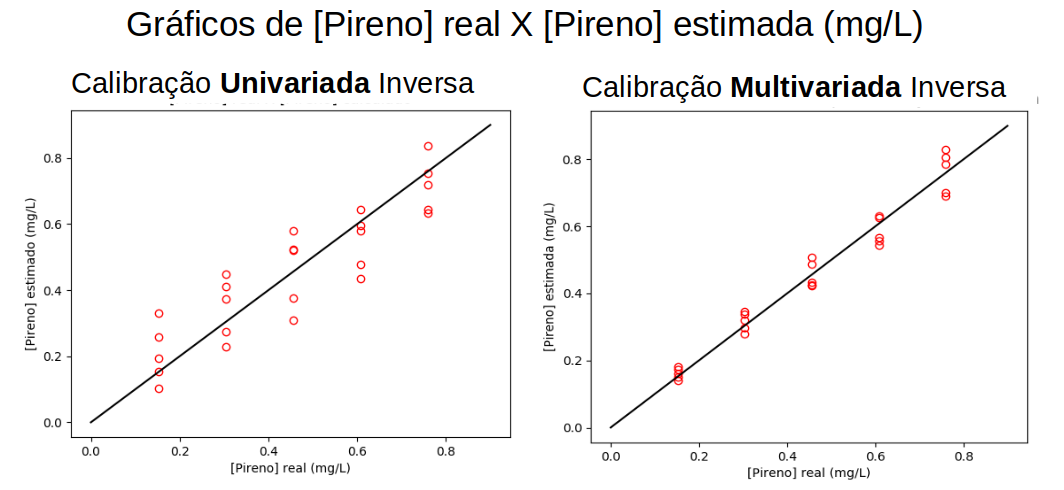
O gráfico da figura V.4 mostra a diferença na dispersão dos resultados obtidos no cálculo da concentração de Pireno quando se utiliza a Calibração Univariada Inversa com 1 comprimento de onda (330nm), e a Calibração Multivariada Inversa baseada em 4 comprimentos de onda (330, 335, 340 e 345nm).
Figura V.4. À esquerda o gráfico das concentrações estimadas de pireno em relação às concentrações reais, obtidas pela técnica de Calibração Univariada Inversa com 1 comprimento de onda (330nm); e à direita o gráfico das concentrações estimadas de pireno em relação às concentrações reais, obtidas pela técnica de Calibração Multivariada Inversa utilizando 4 comprimentos de onda (330, 335, 340 e 345nm) para a calibração.
Nota
Os comandos usados nesta seção estão organizados em um único arquivo disponível no link: regressao_multivariada_00.py.